Dashboard Components¶
Depictio provides a variety of component types for building interactive dashboards. This guide covers each component type, when to use it, and how to configure it.
Browse the live catalog
To see which bioinformatics tool outputs map to these components, explore the interactive Depictio Modules gallery — a live, offline catalog of tool-to-component integrations.
Component Overview¶
-
Charts and plots (scatter, bar, histogram, line, box, pie, and more)
-
Interactive data tables with filtering and sorting
-
Metric display with aggregations
-
Section headers (H1, H2, H3)
-
User input components for filtering (slider, dropdown, date picker)
-
Quality control report visualizations
-
Image galleries with S3/MinIO storage integration
-
Geospatial map visualization with markers
-
Domain-specific scientific viz (volcano, MA, manhattan, ComplexHeatmap, UpSet, sankey, …) backed by canonical column schemas
Figure Components (v0.0.1+)¶
Figure components display data visualizations using Plotly charts. They support both UI Mode (drag-and-drop configuration) and Code Mode (Python code for custom plots).
Supported Chart Types¶
UI Mode provides a curated selection of chart types through the visual interface:
| Category | Chart Types |
|---|---|
| Basic Charts | Scatter, Bar, Line |
| Statistical | Histogram, Box |
| Matrix | ComplexHeatmap (via plotly-complexheatmap) |
Unlimited Charts with Code Mode
Code Mode supports the entire Plotly library, giving you access to all chart types including 3D plots, maps, financial charts, and more. See the Plotly Python documentation for the complete reference.
UI Mode¶
In UI Mode, configure charts through the visual interface:
- Select a Data Collection as your data source
- Choose the Chart Type (scatter, bar, histogram, etc.)
- Map data columns to X-axis, Y-axis, and optional Color dimension
- Customize appearance (title, axis labels, colors)
Code Mode¶
Code Mode allows custom Python code for advanced visualizations:
import plotly.express as px
# df is your data collection as a pandas DataFrame
fig = px.scatter(
df,
x="coverage",
y="quality_score",
color="sample_type",
title="Coverage vs Quality by Sample Type"
)
# Return the figure object
fig
Security Note
Code Mode uses RestrictedPython for security. Only approved libraries (pandas, plotly) are available. See Security for details.
Configuration Options¶
| Option | Description | Default |
|---|---|---|
| Title | Chart title displayed at top | Auto-generated |
| X-axis label | Label for horizontal axis | Column name |
| Y-axis label | Label for vertical axis | Column name |
| Color | Column for color encoding | None |
| Hover data | Additional columns shown on hover | None |
Clustered heatmaps moved
The ComplexHeatmap viz has been renamed and is now part of the Advanced Visualizations section below — see Hierarchical Heatmap for the full config, alongside the rest of the domain-specific viz family (volcano, MA, sankey, …).
Selection Filtering (Scatter Plots)¶
Scatter plots can act as interactive filters. Enable selection to let users lasso, box-select, or click points to filter other components.
| Option | Description |
|---|---|
selection_enabled |
Enable selection filtering (true/false) |
selection_column |
Column to extract from selected points |
See Interactive Selection Filtering for details.
Table Components (v0.0.2+)¶
Table components display data in interactive tables with built-in filtering and sorting.
Features¶
| Feature | Description |
|---|---|
| Server-Side Pagination | Efficiently handles large datasets by loading data in pages |
| Server-Side Scrolling | Virtual scrolling for smooth navigation through large tables |
| Column Sorting | Click headers to sort ascending/descending |
| Column Filtering | Filter by column values |
| Export (v0.6.0+) | Download data as CSV |
Configuration¶
| Option | Description | Default |
|---|---|---|
| Data Collection | Source data for the table | Required |
| Visible Columns | Columns to display | All columns |
| Page Size | Rows per page | 10, 25, 50, or 100 |
| Style | Column width, text alignment | Auto |
| Title | Header text above the table | Auto (from DC tag) |
| Description | Subtitle text below the title | None |
| Title Size | Header level: h1, h2, h3, or sm |
sm |
| Title Align | Text alignment: left, center, or right |
left |
Row Selection Filtering¶
Tables can act as interactive filters. Enable row selection to let users click rows to filter other components.
| Option | Description |
|---|---|
row_selection_enabled |
Enable row selection filtering (true/false) |
row_selection_column |
Column to extract from selected rows |
See Interactive Selection Filtering for details.
Advanced Visualizations (v0.13.0+)¶
Advanced Visualizations are a family of domain-specific scientific charts (catalogued below). Unlike the generic Figure component, which accepts any numeric columns, each advanced viz declares a canonical column-role schema: a volcano plot knows it needs a feature_id, an effect_size, and a significance column — wired to your DC's actual column names through the viz's config.
Two ingredients work together:
- a per-viz
Configclass (e.g.VolcanoConfig,MAConfig) that captures the role → column mapping plus per-viz display defaults (thresholds, top-N, sort order); - a
CANONICAL_SCHEMASentry declaring the required and optional roles plus their accepted polars dtypes, used by the dashboard builder to validate the binding and surface errors before the viz renders.
Source of truth in the codebase:
depictio/models/components/advanced_viz/configs.py— per-viz Pydantic configsdepictio/models/components/advanced_viz/schemas.py—CANONICAL_SCHEMAS+validate_binding()depictio/models/components/types.py—AdvancedVizKindliteral enum
Automatic DC recognition (v0.13.10+)¶
When a file is registered as a Data Collection, Depictio scans its column set against a library of producer fingerprints — named patterns that recognise the output of common upstream tools or standard tidy-table shapes. When a fingerprint matches, the dashboard builder surfaces a "Looks like X" hint and pre-selects the matching visualization type in the add-component wizard, saving manual column-binding work.
The four fingerprints added in v0.13.10 cover the metagenomics / ampliseq family:
| Fingerprint name | Detected columns | Suggested viz types |
|---|---|---|
taxonomy_levels_long |
Taxonomic rank columns (e.g. Kingdom, Phylum, …, Genus/Species) + an abundance / fraction column | Sankey, Sunburst |
rarefaction_iter_long |
sample_id, a sequencing-depth column, an iteration column, and ≥1 alpha-diversity metric |
Rarefaction |
alpha_diversity_wide |
sample_id, shannon (or shannon_entropy), observed_features, evenness |
Advanced viz > alpha diversity |
taxonomy_abundance_long |
sample_id, a taxonomy column, a relative-abundance column |
ComplexHeatmap, Stacked taxonomy, Sunburst |
Fingerprint matching is additive: a single DC can match several fingerprints (e.g. a long taxonomy table with per-iteration depth columns would match both taxonomy_levels_long and rarefaction_iter_long). Unmatched DCs behave as before — all viz types remain available for manual binding.
Source of truth: depictio/models/components/advanced_viz/producer_fingerprints.py.
Catalog¶
Every advanced viz consumes a tabular DC (CSV / TSV / Parquet → polars) with the column roles documented per-viz below. The Accepted input column lists upstream tools whose output natively has — or trivially reshapes to — those columns. Web renderers that look like a viz (Microreact, iTOL, IGV, JBrowse, Krona, EnhancedVolcano, qqman, …) aren't listed here — they're peers, not data sources; we call them out in the per-viz prose where the framing helps.
| Viz | Description | Accepted input (canonical producer) |
|---|---|---|
| Volcano | Effect size vs significance scatter for differential analysis. | DESeq2 results table (results() → TSV) |
| MA | Mean intensity vs log fold change for DE / proteomics QC. | DESeq2 results table: log2(baseMean+1) (or log10) → avg_log_intensity, log2FoldChange straight |
| DA barplot | Ranked signed-LFC bars for differential abundance — single panel or faceted by contrast. | ANCOM-BC output$res (feature, contrast, lfc, q-value) |
| Enrichment | Pathway / GO-term enrichment dot plot. | clusterProfiler GSEA / ORA result (term, NES, padj, gene-count) |
| Manhattan | Genome-wide signal scatter across chromosomes (GWAS). | PLINK .assoc (chr, pos, p-value) |
| Lollipop | Variant / mutation track along a gene body. | maftools / vcf2maf Mutation Annotation Format table — Hugo_Symbol, Start_Position, Variant_Classification (file format, not Minor Allele Frequency) |
| Coverage track | Read depth / signal along genomic coordinates. | mosdepth per-base / by-region BED (chrom, pos, depth) |
| Stacked taxonomy | Per-sample relative-abundance composition by taxonomic rank. | QIIME2 taxa-collapse table (sample × taxon abundance) |
| Sunburst | Hierarchical taxonomy / pathway viewer. | Kraken2 .kreport parsed into rank columns + fraction_total_reads (Bracken .bracken is flat single-rank, needs lineage expansion first) |
| Rarefaction | Alpha-diversity vs sequencing-depth saturation curve. | QIIME2 alpha-rarefaction (sample, depth, alpha-metric) |
| Phylogenetic | Newick tree + tip metadata. Renders Microreact-style. | IQ-TREE Newick + a tabular tip-metadata TSV |
| Dot plot | Single-cell marker-gene expression by cluster. | scanpy aggregation (sc.get.aggregate or groupby on adata.X) producing (cluster, gene, mean_expression, frac_expressing) — rank_genes_groups.to_df() alone is DE stats, not the dot-plot schema |
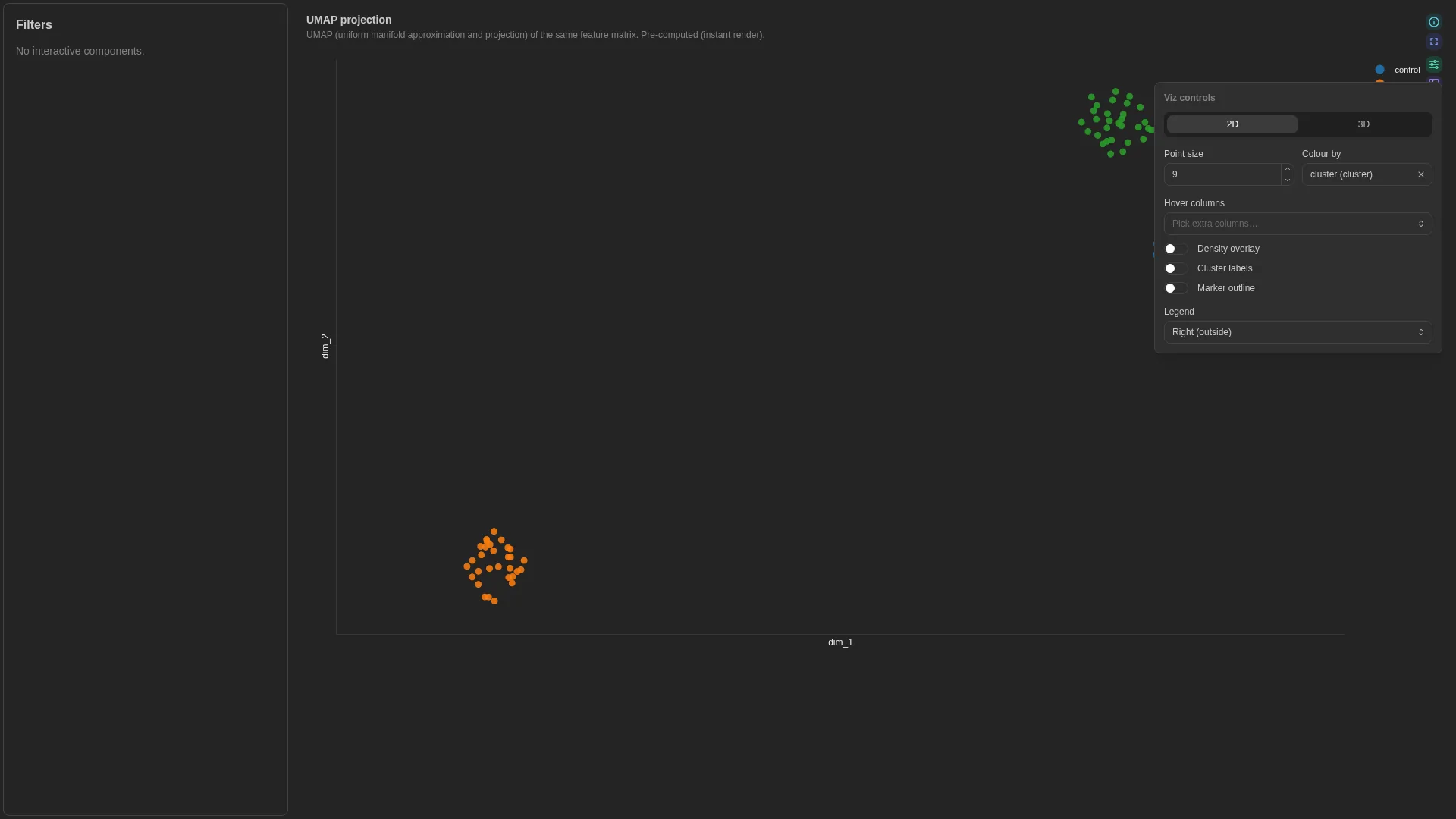
| Embedding | 2D / 3D sample projection for cluster inspection (precomputed or live PCA / UMAP / t-SNE / PCoA). | scanpy adata.obsm['X_umap'] (sample, dim1, dim2) |
| Hierarchical Heatmap | Clustered matrix with dendrograms + annotation tracks. | DESeq2 vst() matrix (sample × gene wide) |
| p-value distribution QC for inflation / deflation. | PLINK .assoc (or any p-value column) |
|
| UpSet | Set-intersection visualisation, alternative to Venn. | Any binary membership matrix (sample × set) |
| Sankey | Categorical flow across N ordered levels. | Any tidy table with ≥2 ordered categorical columns |
| Oncoplot | Sample × gene mutation matrix. | maftools / vcf2maf Mutation Annotation Format table — Tumor_Sample_Barcode, Hugo_Symbol, Variant_Classification (file format, not Minor Allele Frequency) |
Reading the schema tables
Each viz subsection lists its required column roles (must be bound for the viz to render) and optional roles (extra colour / size / label dimensions). Types use polars dtype families — Float accepts Float32 / Float64, Int accepts Int8–Int64 and unsigned widths, String accepts String / Utf8, Numeric is Int ∪ Float. The dashboard builder validates the binding via validate_binding() and surfaces dtype mismatches in-place.
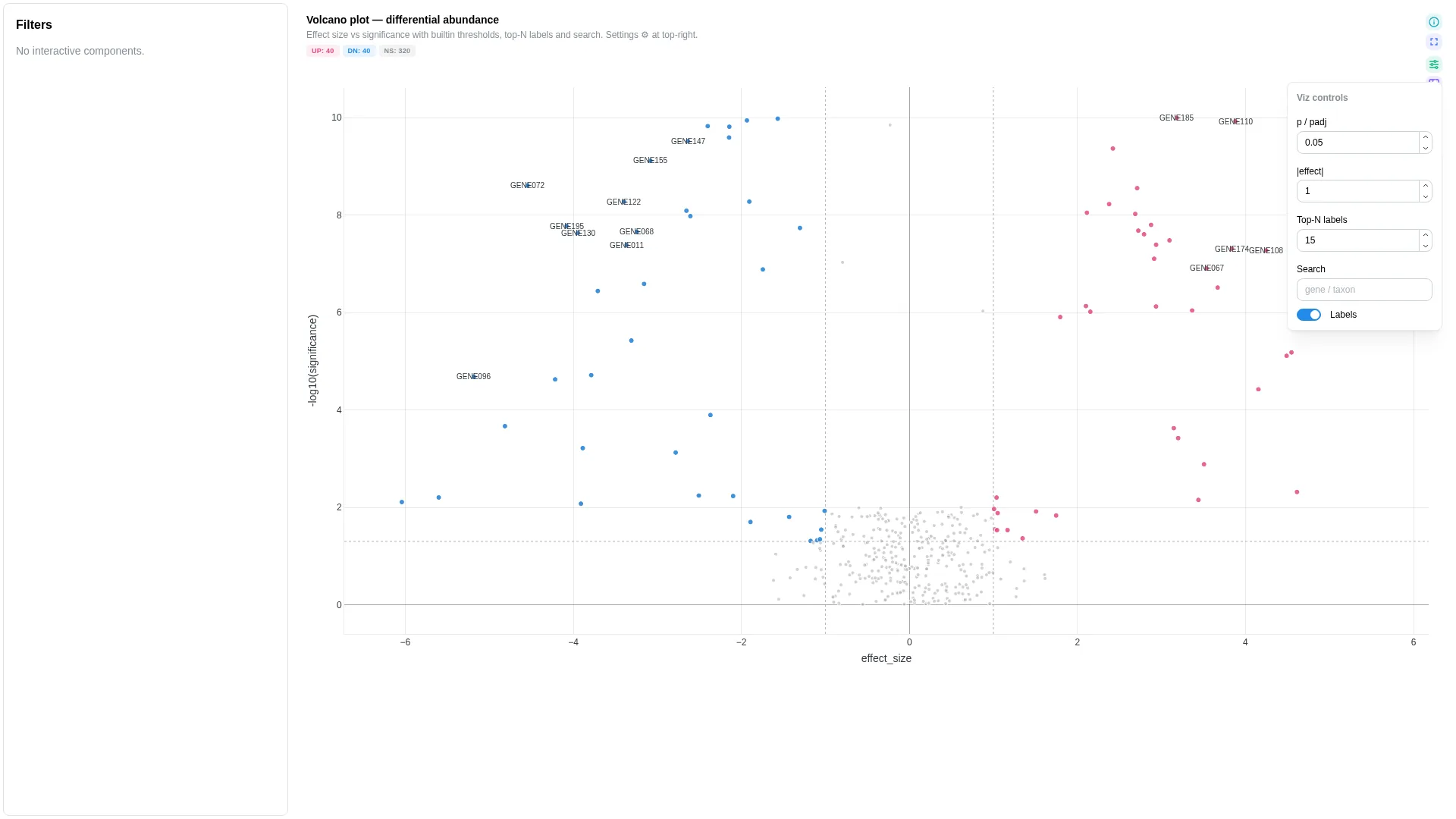
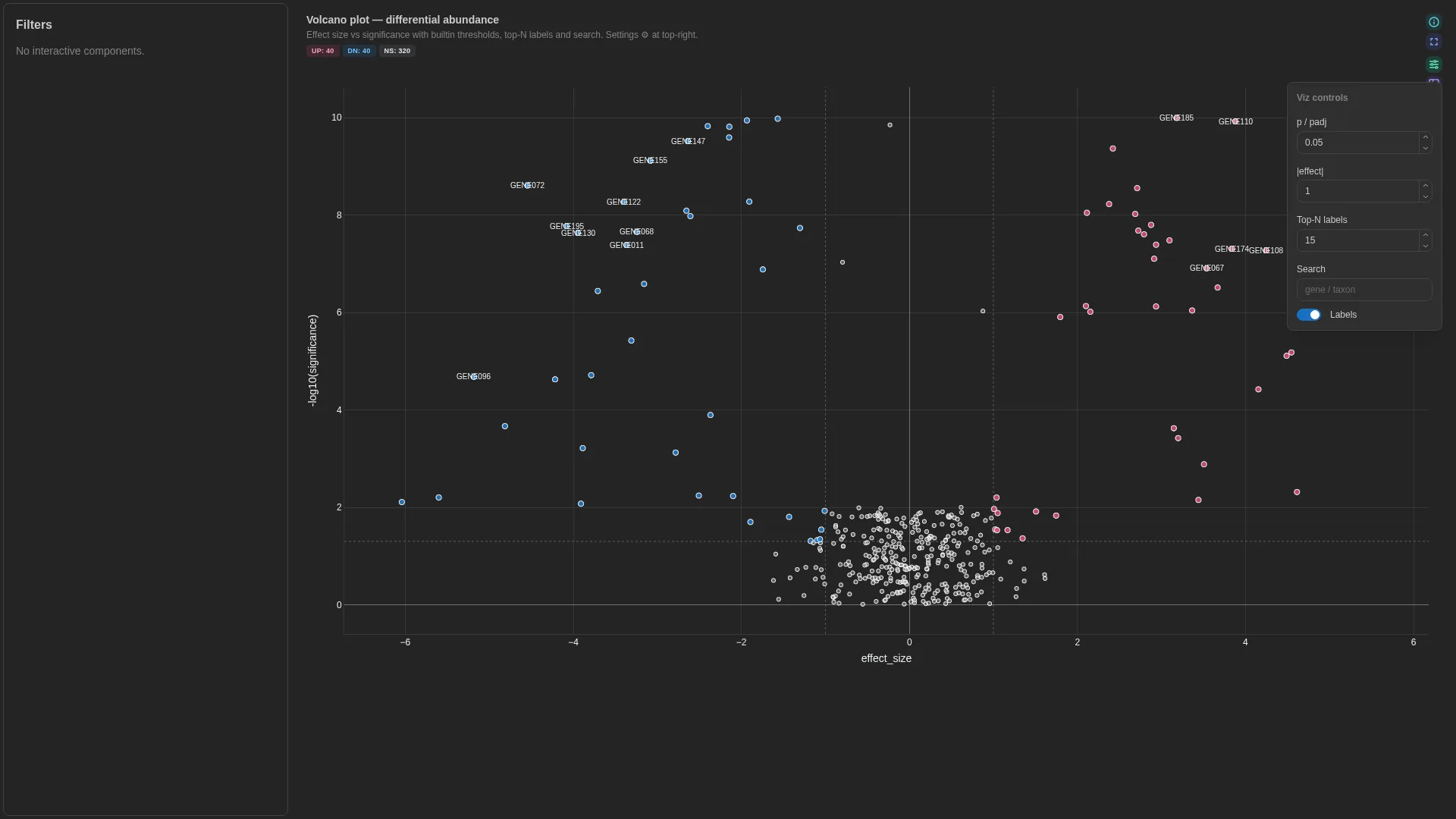
Volcano¶
Effect size vs significance scatter — classic differential-expression view with threshold lines, point search, and top-N labels.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
feature_id |
✓ | String | Feature identifier (gene, peak, …) |
effect_size |
✓ | Float | Effect size (e.g. log2FC, lfc) |
significance |
✓ | Float | p-value or padj/q-value |
label |
— | String | Hover label override |
category |
— | String | Categorical annotation (pathway, cluster…) for point colour |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
significance_is_neg_log10 |
bool | false |
True if significance_col already contains -log10(p); else applied client-side |
significance_threshold |
float | 0.05 |
Cutoff applied to the significance column |
effect_threshold |
float | 1.0 |
Absolute effect_size cutoff |
top_n_labels |
int (≥0) | 20 |
Max features to auto-label |
Filtering / row tagging
Every row is classified client-side as UP, DOWN, or NS based on significance < threshold combined with |effect_size| > threshold. The backend returns raw rows; classification + colouring happens in VolcanoRenderer.tsx.
MA¶
Mean log intensity (x) vs log2 fold change (y) — same hits as volcano, classic DE / proteomics layout. Shares the UP / DOWN / NS tier scheme with Volcano.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
feature_id |
✓ | String | Feature identifier |
avg_log_intensity |
✓ | Float | Mean log intensity (A in MA, x-axis). For DESeq2: pre-transform baseMean with log2(baseMean + 1) (or log10) — baseMean itself is untransformed normalised counts. |
log2_fold_change |
✓ | Float | Log2 fold change (M in MA, y-axis). DESeq2 log2FoldChange maps directly. |
significance |
— | Float | p / padj column for tier colouring |
label |
— | String | Hover label override |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
significance_threshold |
float (0–1) | 0.05 |
Cutoff for significance |
fold_change_threshold |
float (≥0) | 1.0 |
Absolute log2_fold_change cutoff |
top_n_labels |
int (≥0) | 15 |
Max features to auto-label |
Filtering / row tagging
Mirror of the Volcano tier scheme — UP / DOWN / NS classification (sig × FC thresholds), client-side in MARenderer.tsx.
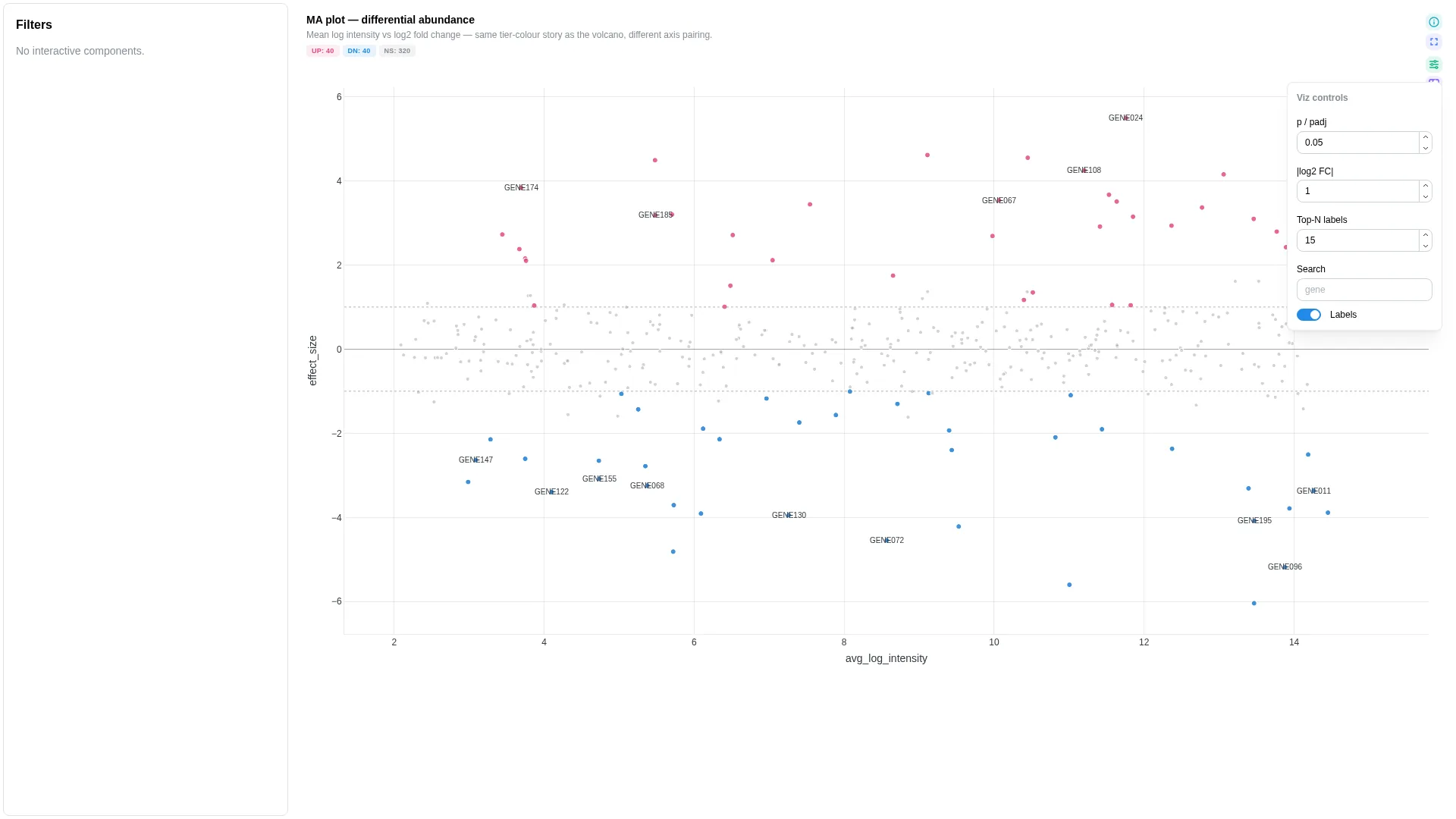
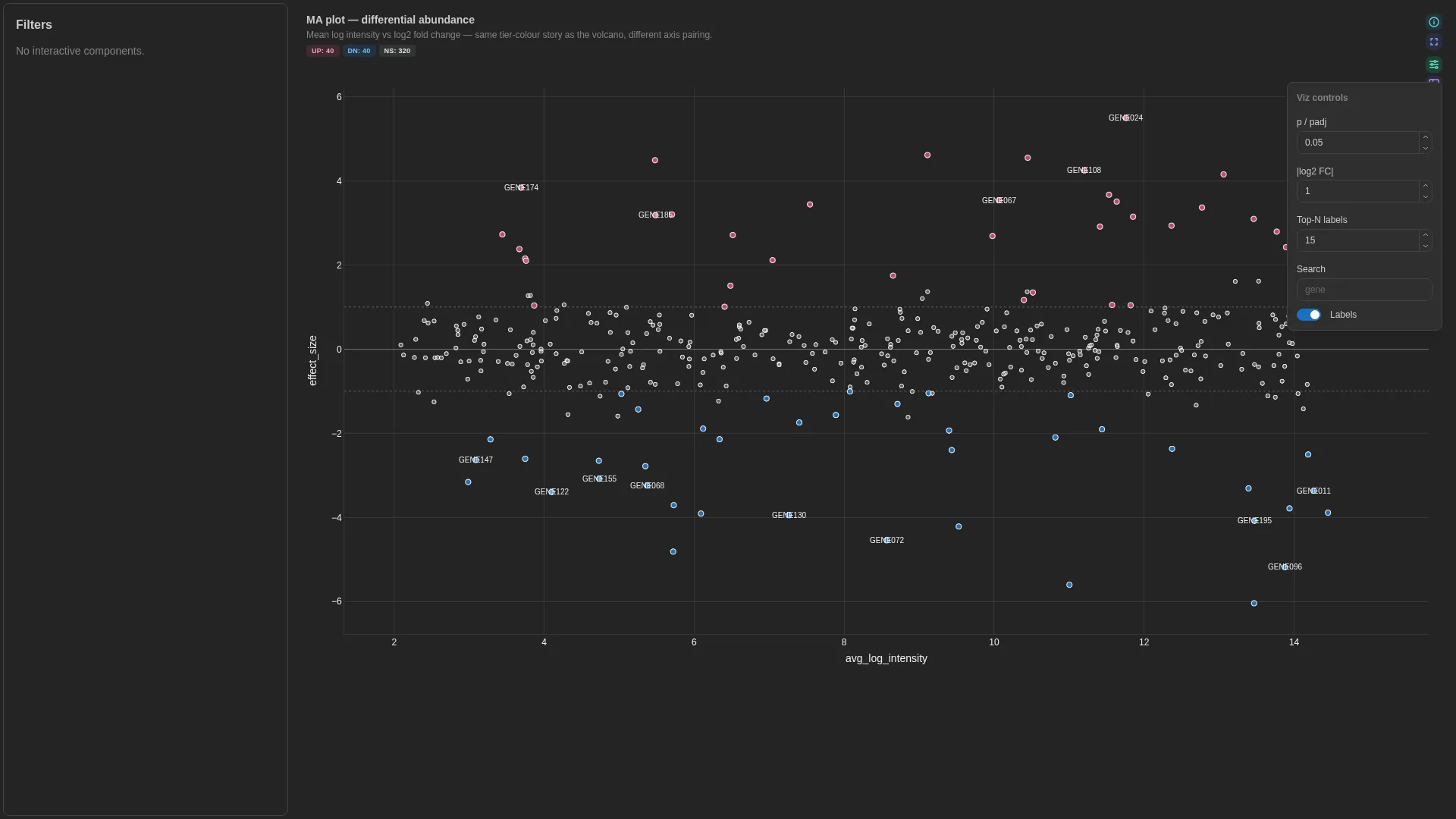
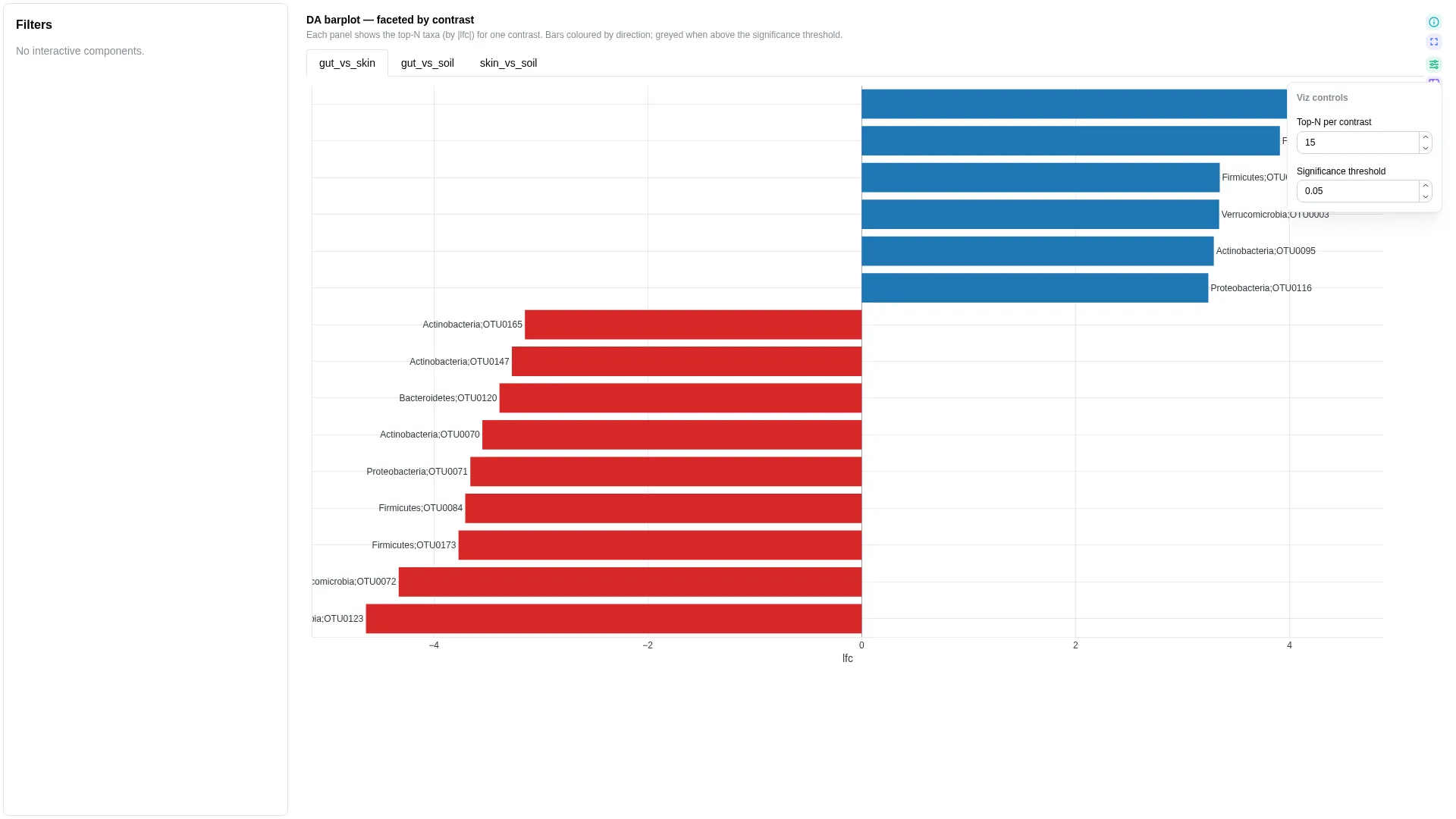
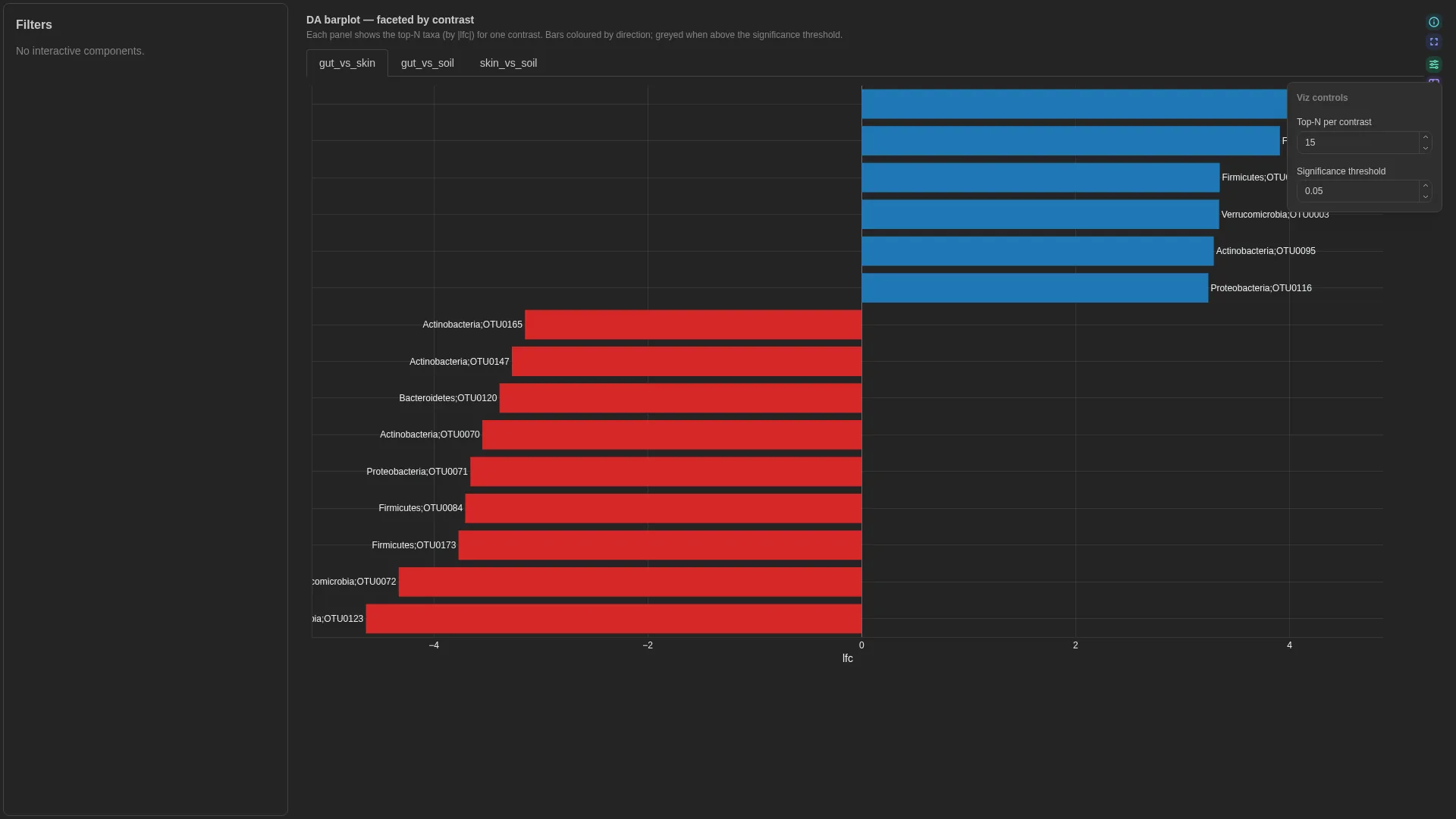
DA barplot¶
Ranked signed-LFC horizontal bars for differential abundance — single panel or faceted across contrasts. Same input shape as the upstream tool (ANCOM-BC, ALDEx2, MaAsLin2): one row per (feature, contrast) with lfc and optional significance.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
feature_id |
✓ | String | Feature / taxon identifier |
contrast |
✓ | String | Contrast name (faceting + single-panel filter) |
lfc |
✓ | Float | Log-fold-change (signed) |
significance |
— | Float | FDR-adjusted p-value; significant bars are highlighted when bound |
label |
— | String | Display label for bars |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
contrast_view |
str | "all" |
"all" → faceted small-multiples (one panel per contrast); any specific contrast value → single-panel drill-in |
significance_threshold |
float (0–1) | 0.05 |
Highlight cutoff for significance |
top_n |
int (≥1) | 15 |
Top features by \|lfc\| shown per panel |
Filtering / row tagging
When contrast_view == "all" the renderer facets by contrast (one panel per unique value, top-N per panel). When contrast_view matches a specific contrast value, the renderer collapses to a single panel showing only that contrast — useful when one comparison is the focus.
Legacy viz_kind: ancombc_differentials
Previously this single-panel layout was a separate viz kind named ancombc_differentials. It's now merged into DA barplot — viz_kind: ancombc_differentials is still accepted at deserialisation and rewritten to da_barplot with contrast_view defaulted from the persisted config.
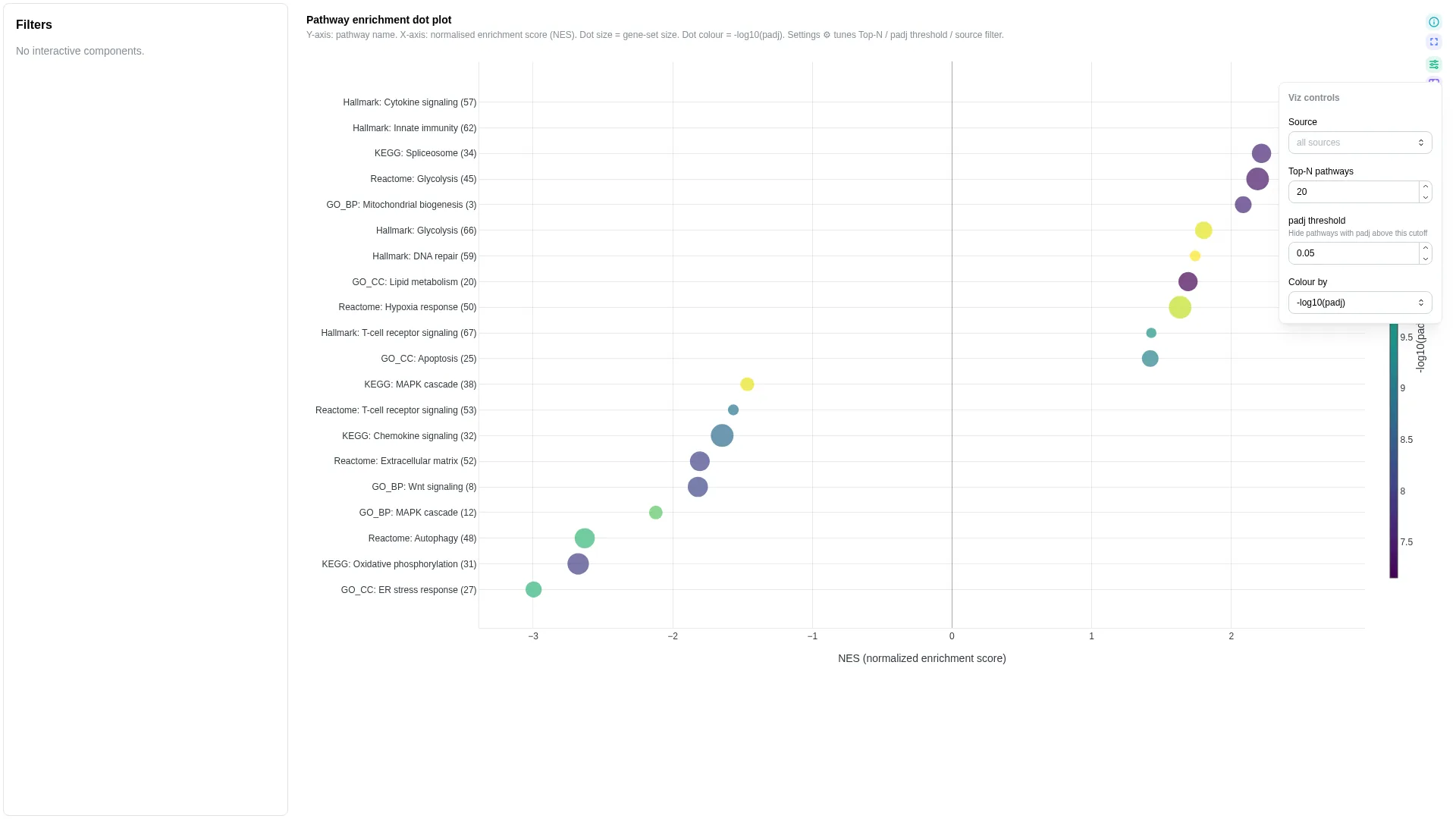
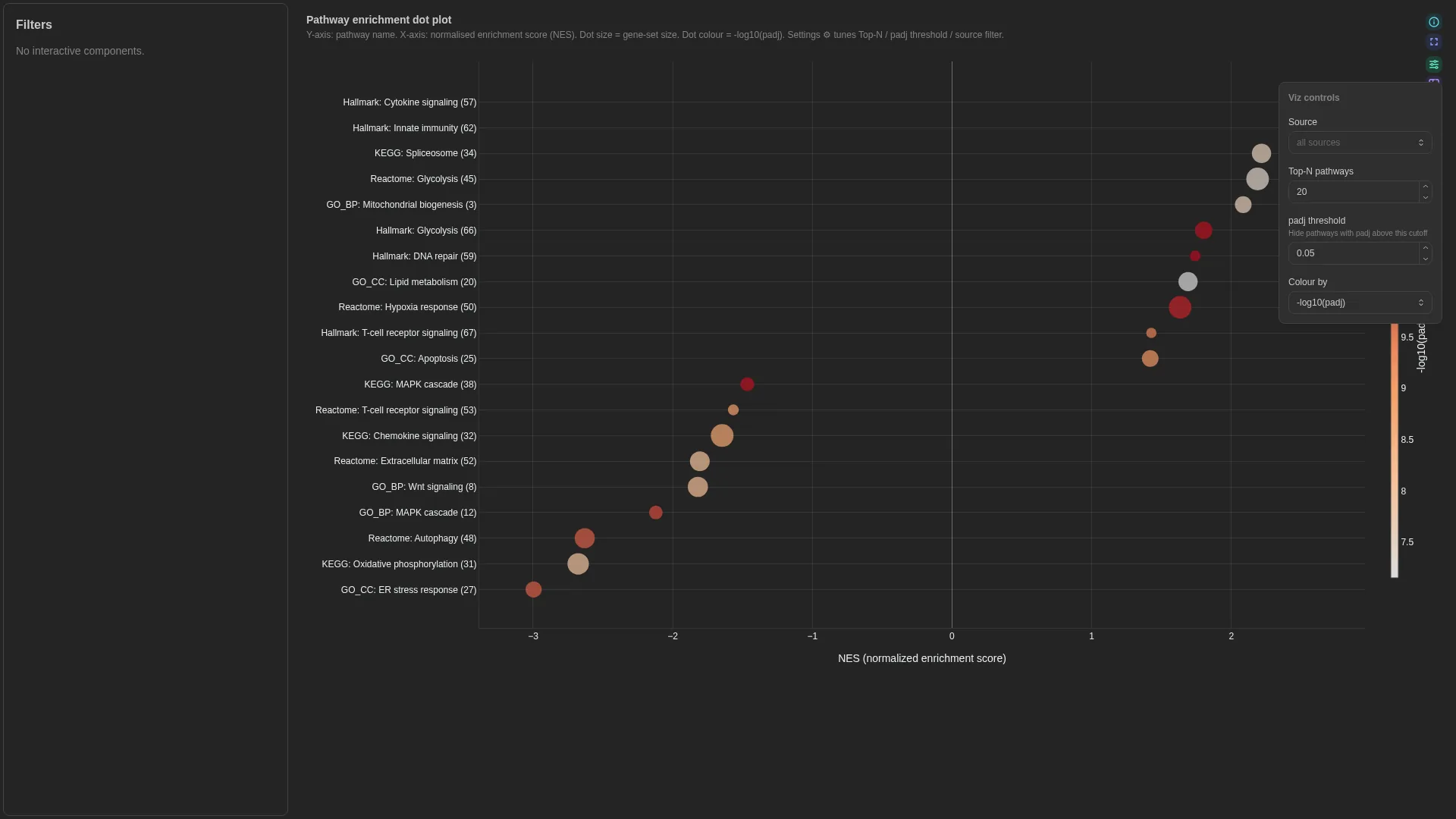
Enrichment¶
GSEA / GO / KEGG / Reactome pathway-enrichment dot plot: term on y, NES on x, dot size = gene-set size, colour = -log10(padj).
Columns
| Role | Required | Type | Description |
|---|---|---|---|
term |
✓ | String | Pathway / GO-term name |
nes |
✓ | Float | Normalised enrichment score (signed, x-axis) |
padj |
✓ | Float | FDR-adjusted p-value |
gene_count |
✓ | Numeric | Gene-set size (dot size) |
source |
— | String | Ontology / source label (GO_BP, KEGG, Reactome, Hallmark, …) |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
padj_threshold |
float (0–1) | 0.05 |
Filter cutoff |
top_n |
int (≥1) | 20 |
Max pathways shown |
Filtering / row tagging
Renderer filters by source (MultiSelect) and ranks by |nes|; only the top-N pathways are shown. Dot colour encodes -log10(padj).
Manhattan¶
Generic chr / pos / score plot — works for true GWAS (variants), peak significance (ATAC/ChIP narrowPeak), or viral variant tracks. score_kind keeps the y-axis label honest.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
chr |
✓ | String | Chromosome label |
pos |
✓ | Int | Genomic position (1-based) |
score |
✓ | Float | Y-axis score (e.g. -log10(padj)) |
feature |
— | String | Feature / locus id (gene, SNP, peak) |
effect |
— | Float | Signed effect for point colouring |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
score_kind |
str | -log10(padj) |
Y-axis label override |
score_threshold |
float | null | null |
Horizontal threshold line; null hides it |
highlight |
above | below | none |
above |
Which side of score_threshold gets emphasised. above colours and enlarges points at/above the threshold (GWAS / consensus-variant default); below inverts it (useful for minority-allele / sub-threshold candidates); none colours both sides equally. No effect when score_threshold is null. |
marker_size_above |
int (1–30) | 6 |
Marker size (px) for points at or above score_threshold. Only used when a threshold is set. |
marker_size_below |
int (1–30) | 4 |
Marker size (px) for sub-threshold points. Lower by default so the eye lands on the hits. |
marker_size_uniform |
int (1–30) | 5 |
Marker size when no threshold is set (uniform sizing). |
color_by_columns |
list[str] | [] |
Extra columns fetched alongside the required roles, exposed in the viz Colour-by dropdown. The renderer auto-detects numeric vs categorical (continuous colorscale vs palette). Chromosome and Score (the y-axis column) are always available without listing them here. Typical viralrecon usage: ['effect', 'lineage', 'sample']. |
default_color_by |
str | null | null |
Initial value for the Colour-by dropdown. Either Chromosome, Score, or one of color_by_columns. Defaults to Chromosome when null. |
Filtering / row tagging
Renderer facets by chromosome (one subplot per unique chr value). The optional score_threshold draws a horizontal cutoff line — no explicit per-row tag, but the line gives a visual significance reference.
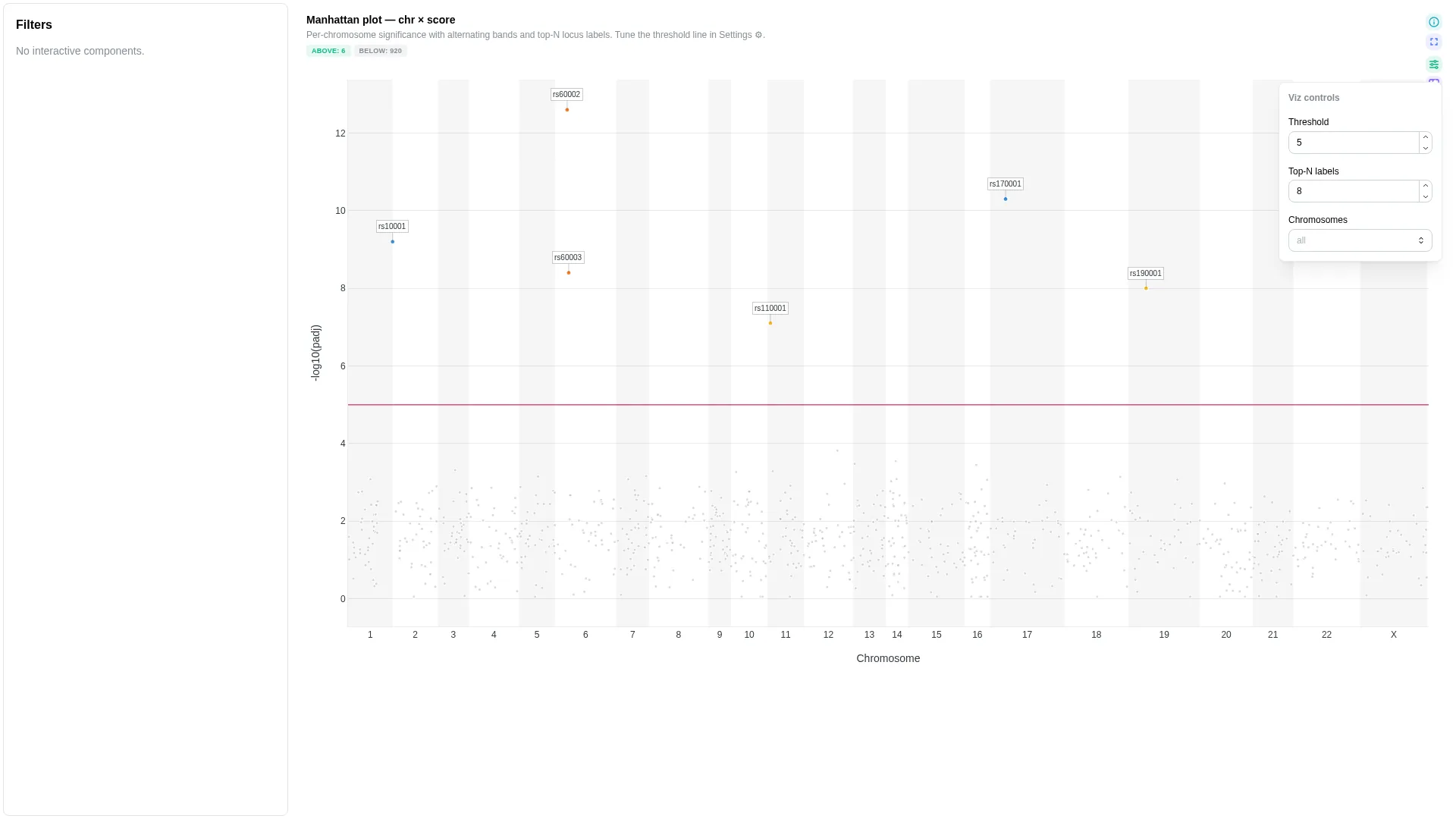
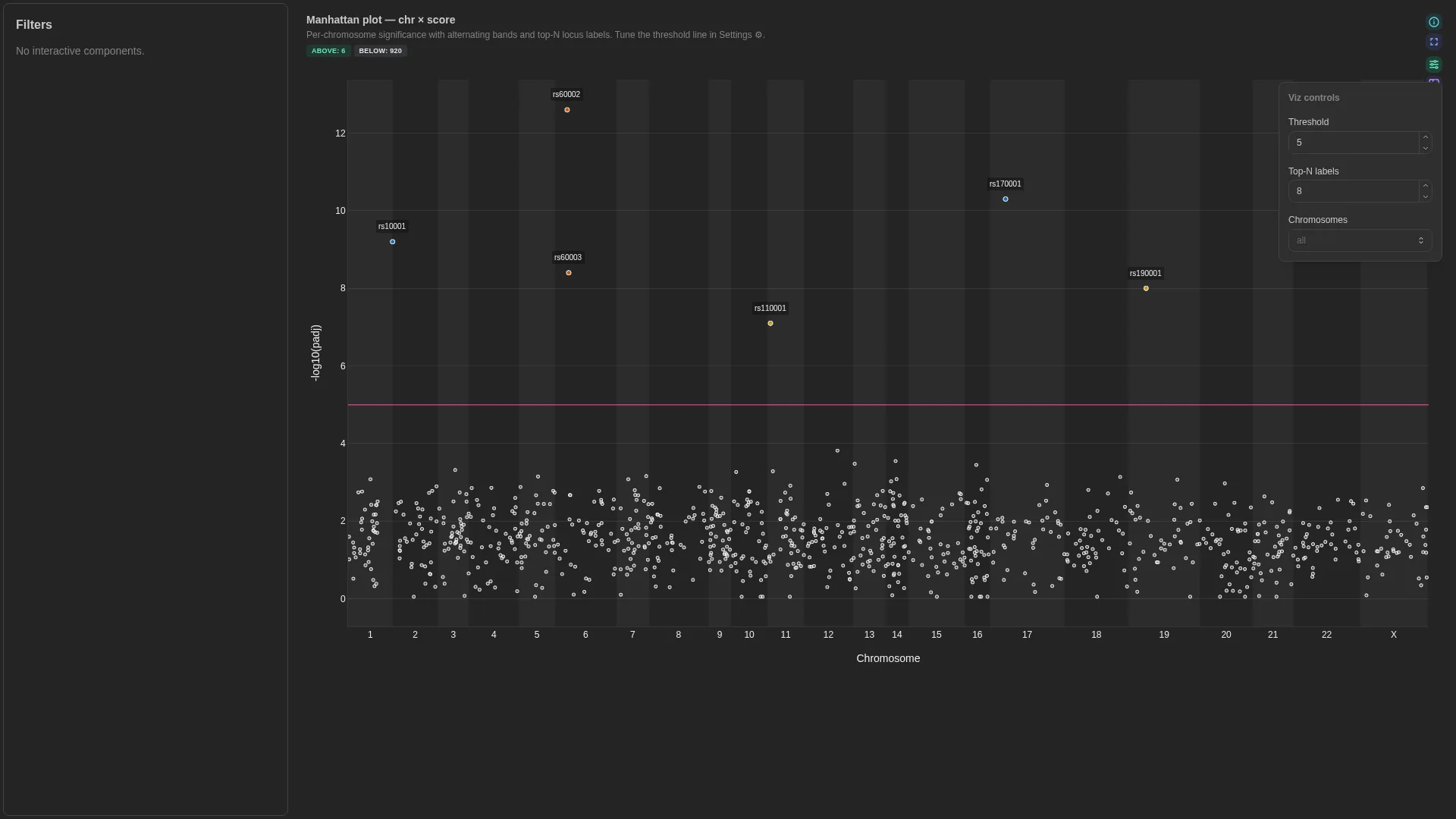
Lollipop¶
Needle / variant track along a gene — each gene body as a horizontal line, each variant as a vertical stem with a category-coloured marker on top.
Columns — straight rename from canonical Mutation Annotation Format (VEP / vcf2maf / maftools — the cancer-mutation file format, not Minor Allele Frequency):
| Role | Required | Type | Description | Mutation Annotation Format column |
|---|---|---|---|---|
feature_id |
✓ | String | Gene / feature the variant is on | Hugo_Symbol |
position |
✓ | Int | Position along the feature | Start_Position (or Protein_position for AA-space tracks) |
category |
✓ | String | Variant consequence category (colour) | Variant_Classification |
effect |
— | Float | Numeric effect (marker size) | e.g. VAF, t_alt_count / t_depth |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
max_subplot_genes |
int (≥1) | 6 |
If the gene universe exceeds this, switch to a single-gene picker |
Filtering / row tagging
Renderer facets by feature (one subplot per gene, or a picker once the universe exceeds max_subplot_genes). Markers are coloured by category and sized by effect when bound.
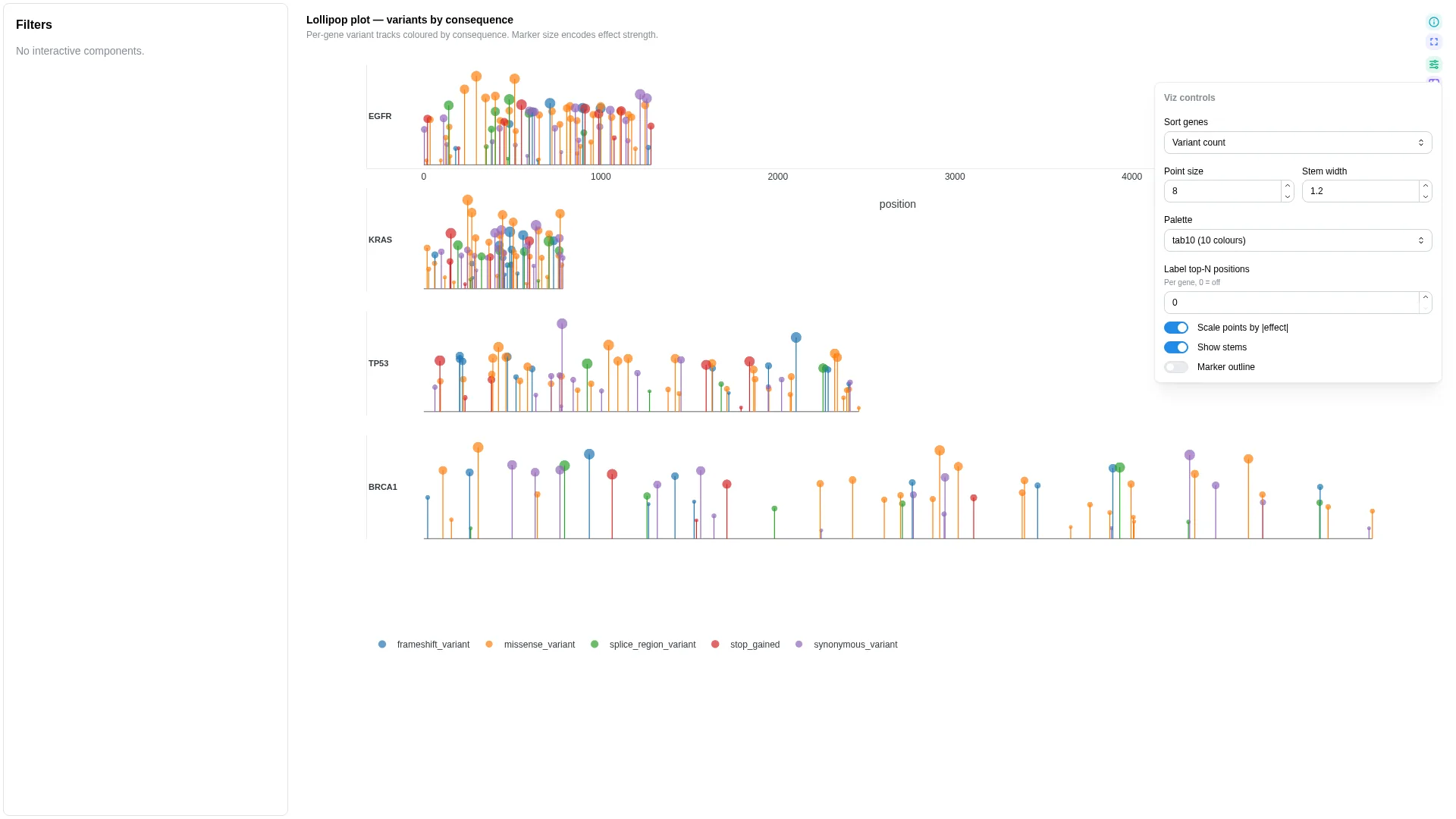
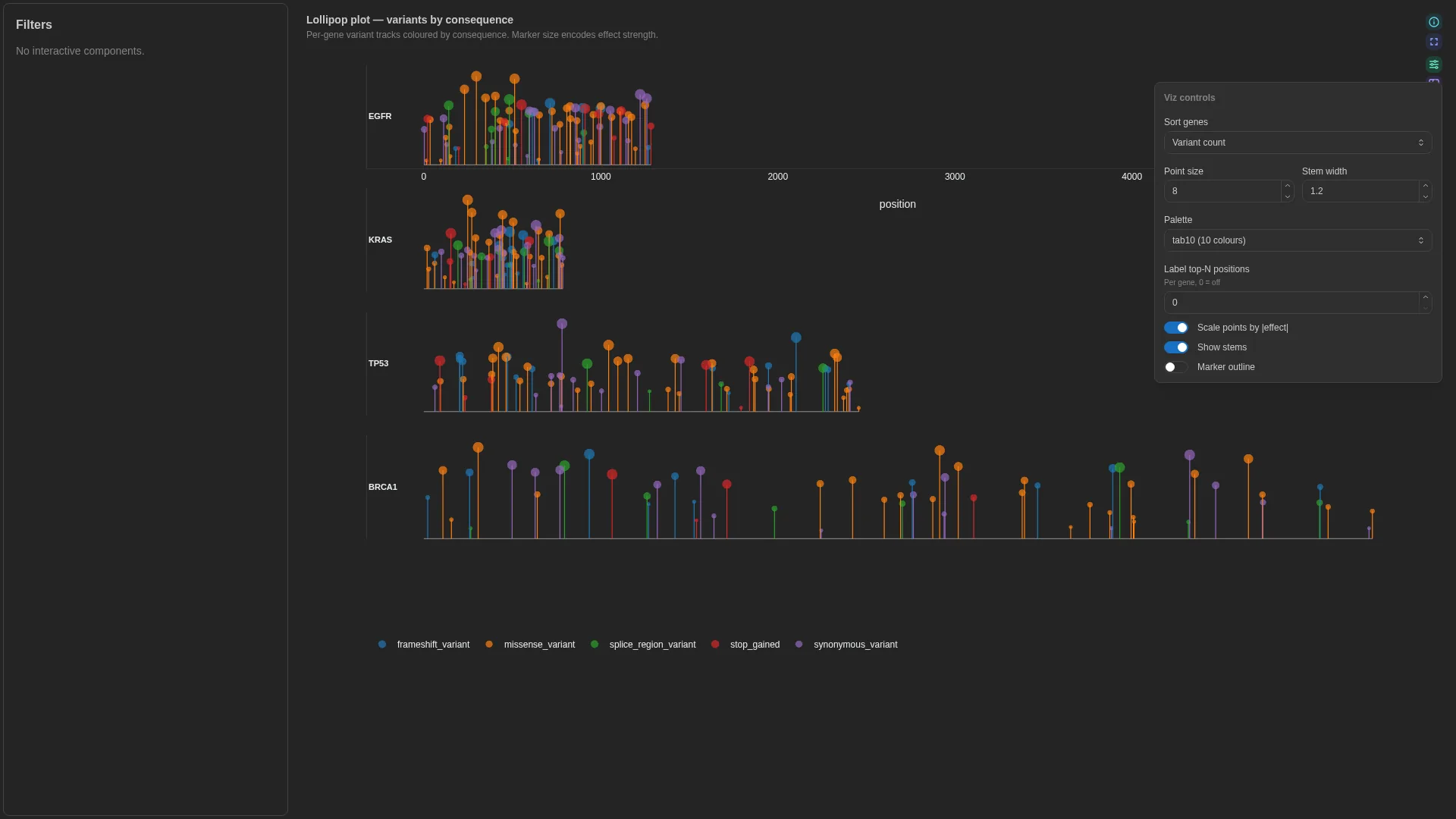
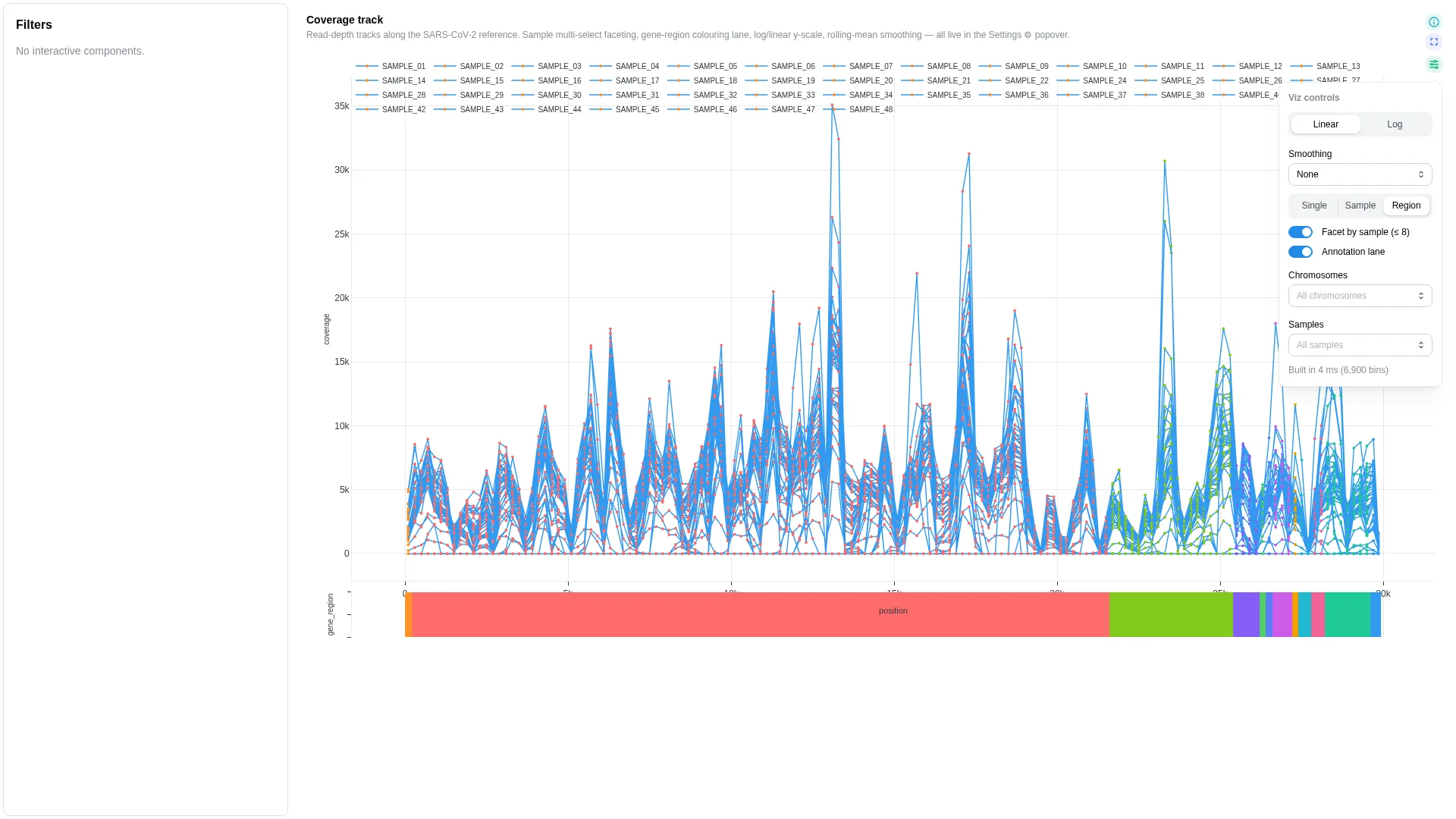
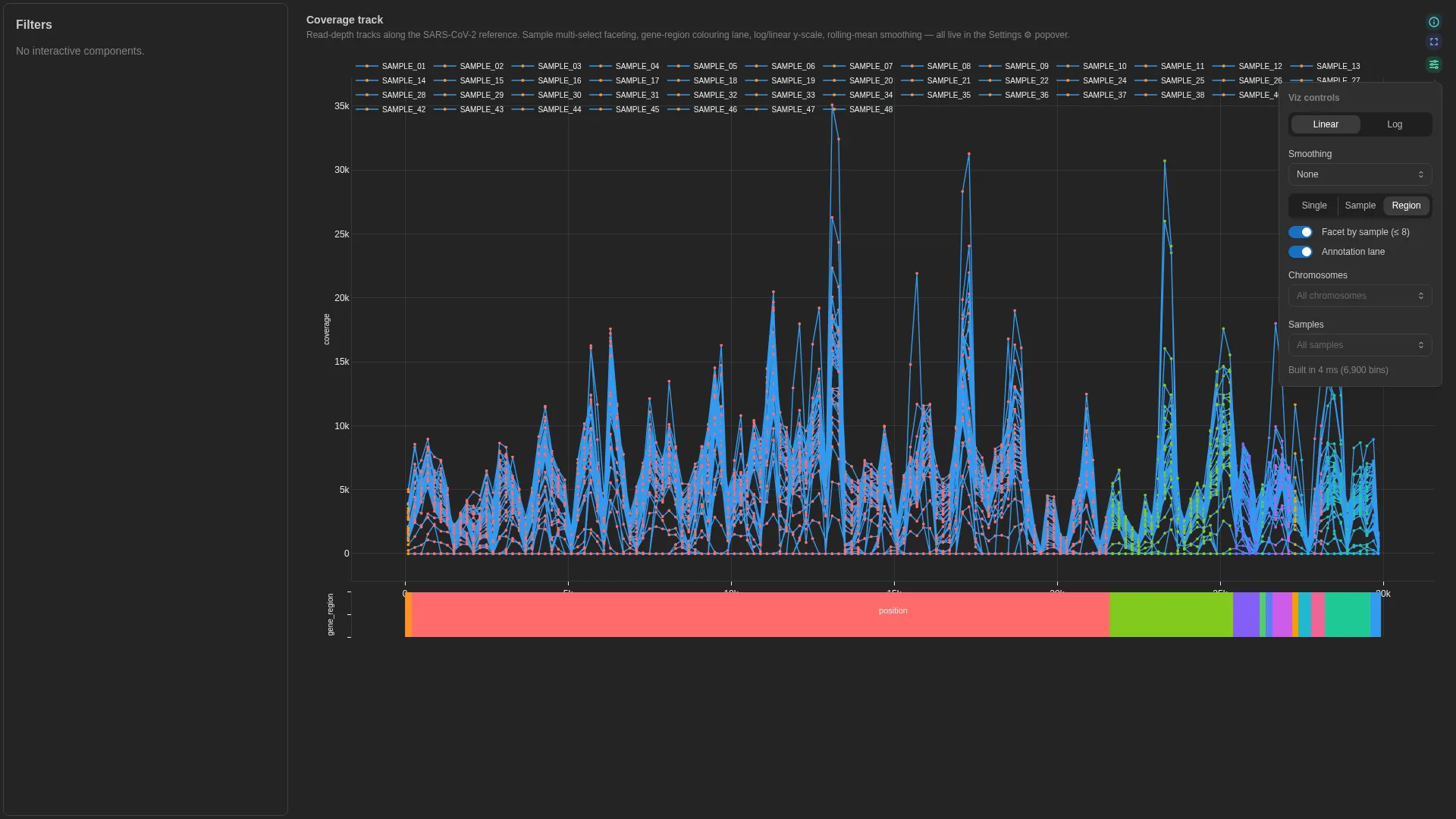
Coverage track¶
Read depth / signal along a coordinate axis. Universal genomics primitive — covers mosdepth bins, BigWig-derived transcript coverage, peak signal, methylseq depth, contig coverage, sarek QC.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
chromosome |
✓ | String | Chromosome / contig label |
position |
✓ | Int | Bin centre or single-base position |
value |
✓ | Numeric | Coverage / signal value |
end |
— | Int | Bin end — when set with position, treated as interval |
sample |
— | String | Per-sample faceting (stacked subplots) |
category |
— | String | Categorical annotation (gene region, peak class, …) |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
y_scale |
linear | log |
linear |
Y-axis scale |
smoothing_window |
int (0–200) | 5 |
Rolling-mean window in bins (0 disables). Default 5 ≈ 1 kb at 200-bp mosdepth bins — kills high-frequency wiggle without flattening amplicon-scale dropouts. |
color_by |
single | category | sample |
single |
Trace colour assignment mode |
show_annotation_lane |
bool | true |
Render annotation strip when category is bound |
annotation_id |
str | null | null |
Optional bundled-annotation override for the genome-feature overlay strip. When null, the renderer auto-detects the assembly from the bound DC's chromosome value (e.g. MN908947.3 → SARS-CoV-2). Pin this when your data uses non-standard chromosome names but corresponds to a known assembly. Valid ids: sars_cov_2, rsv_a, hiv_1, mpox, hbv (see depictio-react-core's genome_annotations registry). |
chromosomes_filter |
list[str] | null | null |
Whitelist of chromosomes; null = all |
samples_filter |
list[str] | null | null |
Whitelist of samples; null = all |
Filtering / row tagging
Renderer facets by chromosome (subplot per chr) and optionally by sample (stacked subplot rows). The chromosomes_filter / samples_filter whitelists narrow the view further; the category lane colour-segments the trace.
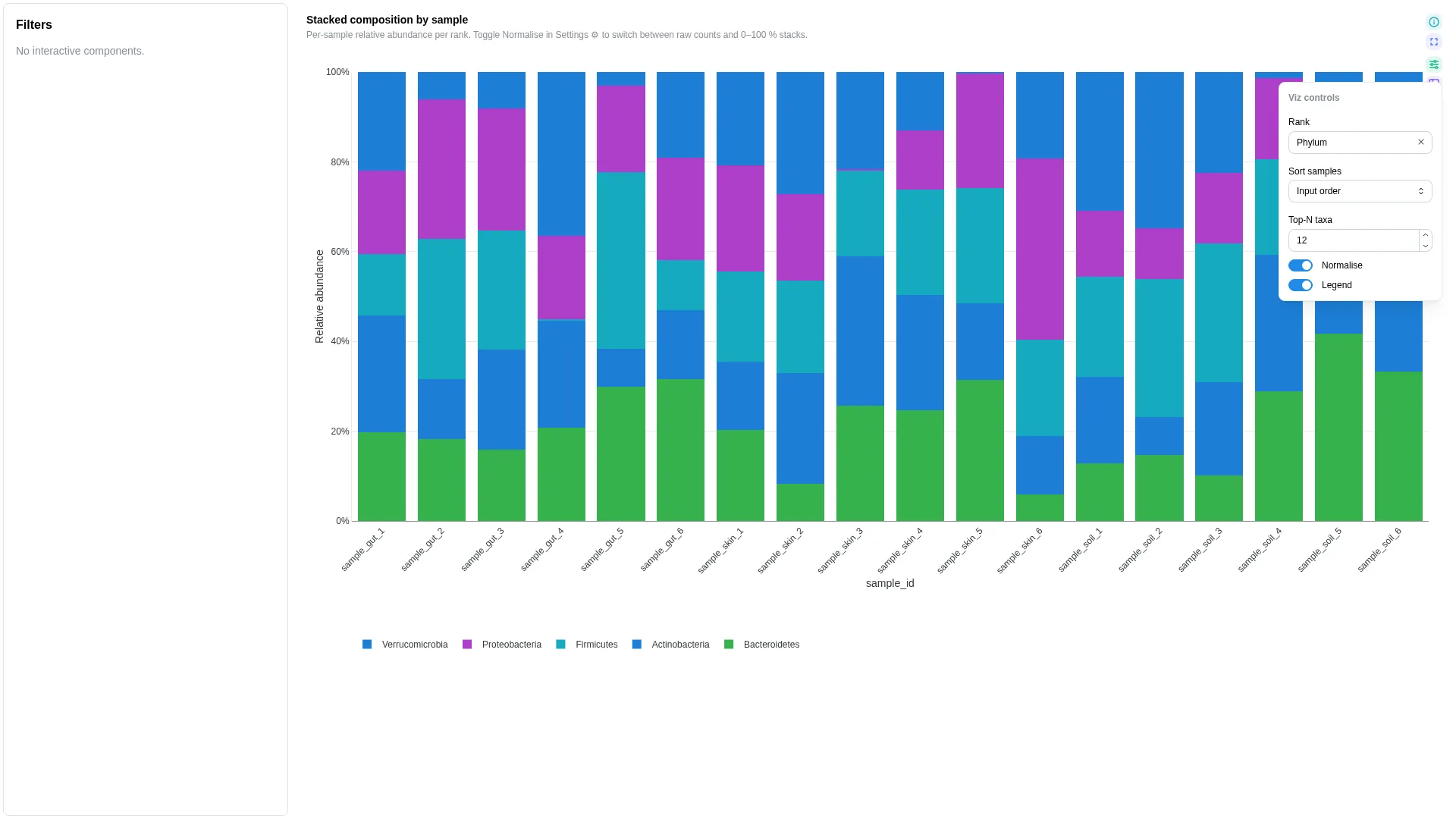
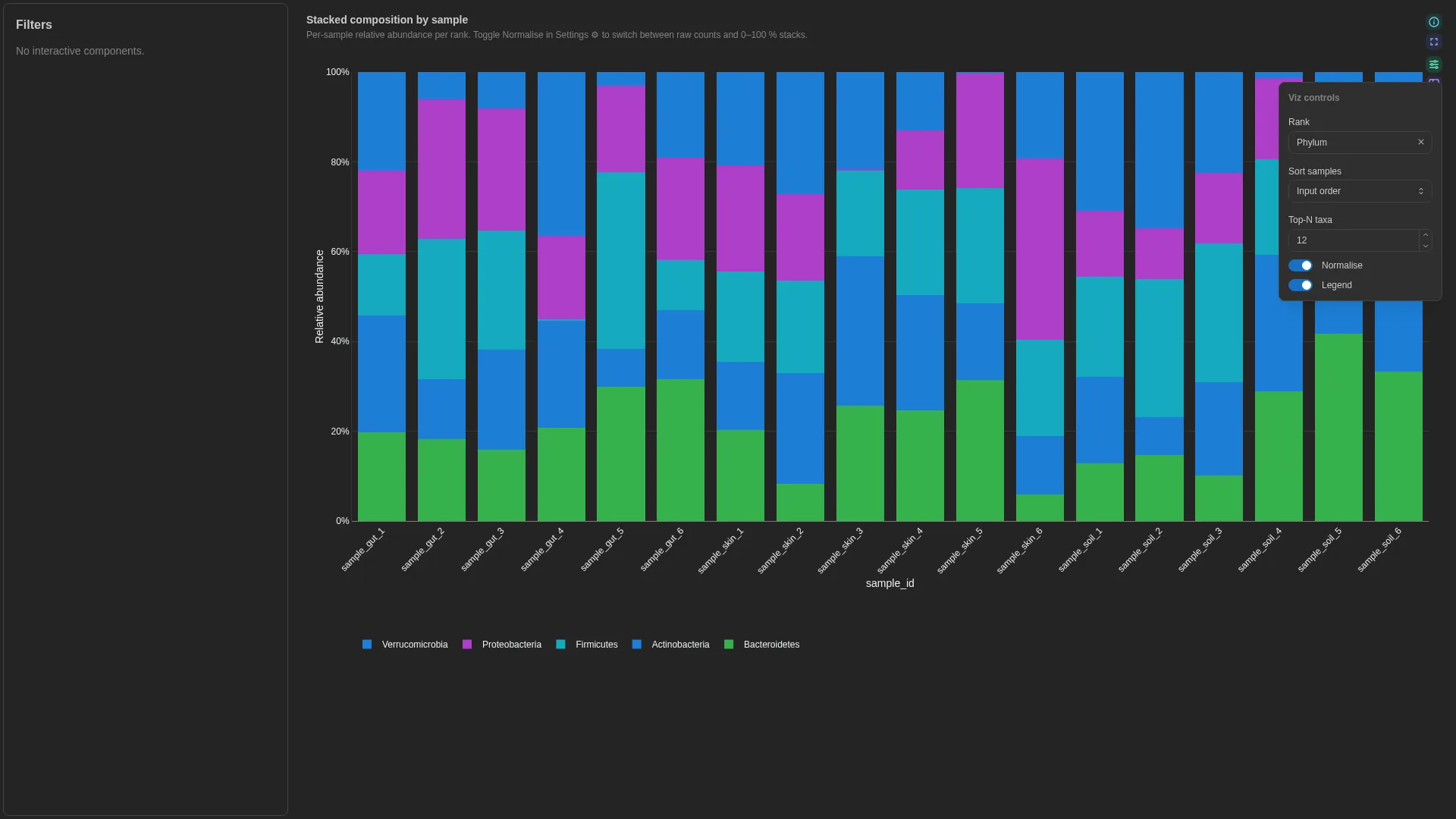
Stacked taxonomy¶
Per-sample stacked relative-abundance bar with a rank dropdown.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
sample_id |
✓ | String | Sample identifier |
taxon |
✓ | String | Taxon name |
rank |
✓ | String | Taxonomic rank label |
abundance |
✓ | Numeric | Relative or absolute abundance |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
default_rank |
str | null | null |
If rank carries multiple ranks, default-filter to this one |
top_n |
int (≥1) | 20 |
Show top-N taxa, lump rest into Other |
sort_by |
abundance | alphabetical |
abundance |
Stack-ordering rule |
normalise_to_one |
bool | true |
Force each sample's bars to sum to 1 (true % composition) |
annotation_strips |
list[dict] | null | null |
Per-sample categorical annotation strips drawn above or below the stacked bars. Each entry is a dict with: column (str, required), label (str, optional — defaults to column name), position (top | bottom, default bottom), palette ({value: hex}, optional). Reusable across any per-sample categorical metadata (habitat, batch, treatment, timepoint) — renderer pulls the columns automatically, no recipe change needed. |
Annotation strips YAML
Filtering / row tagging
Renderer filters by rank (dropdown sourced from the unique rank values). Within the active rank, taxa are sorted by sort_by; everything past top_n is collapsed into an Other slice.
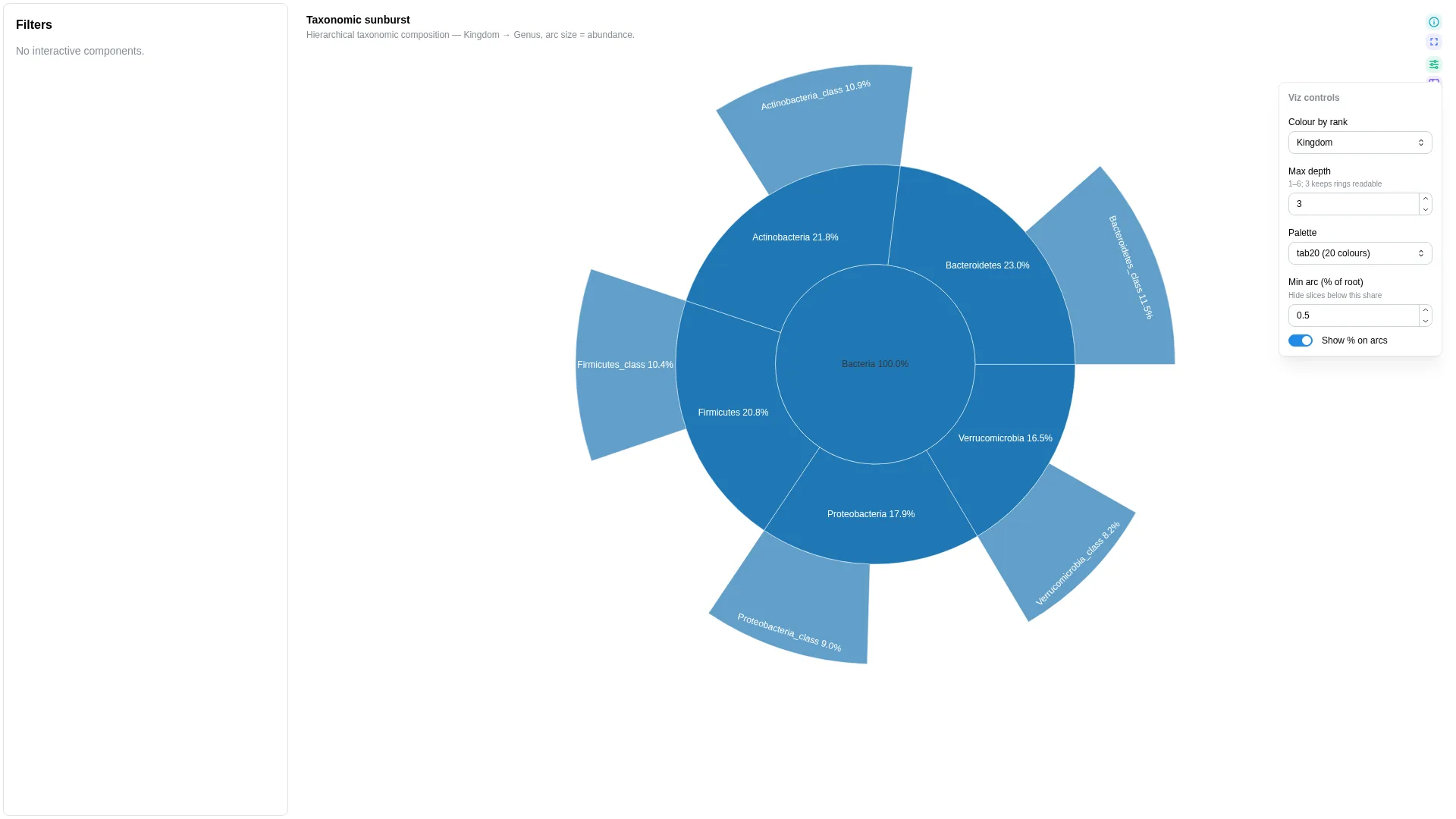
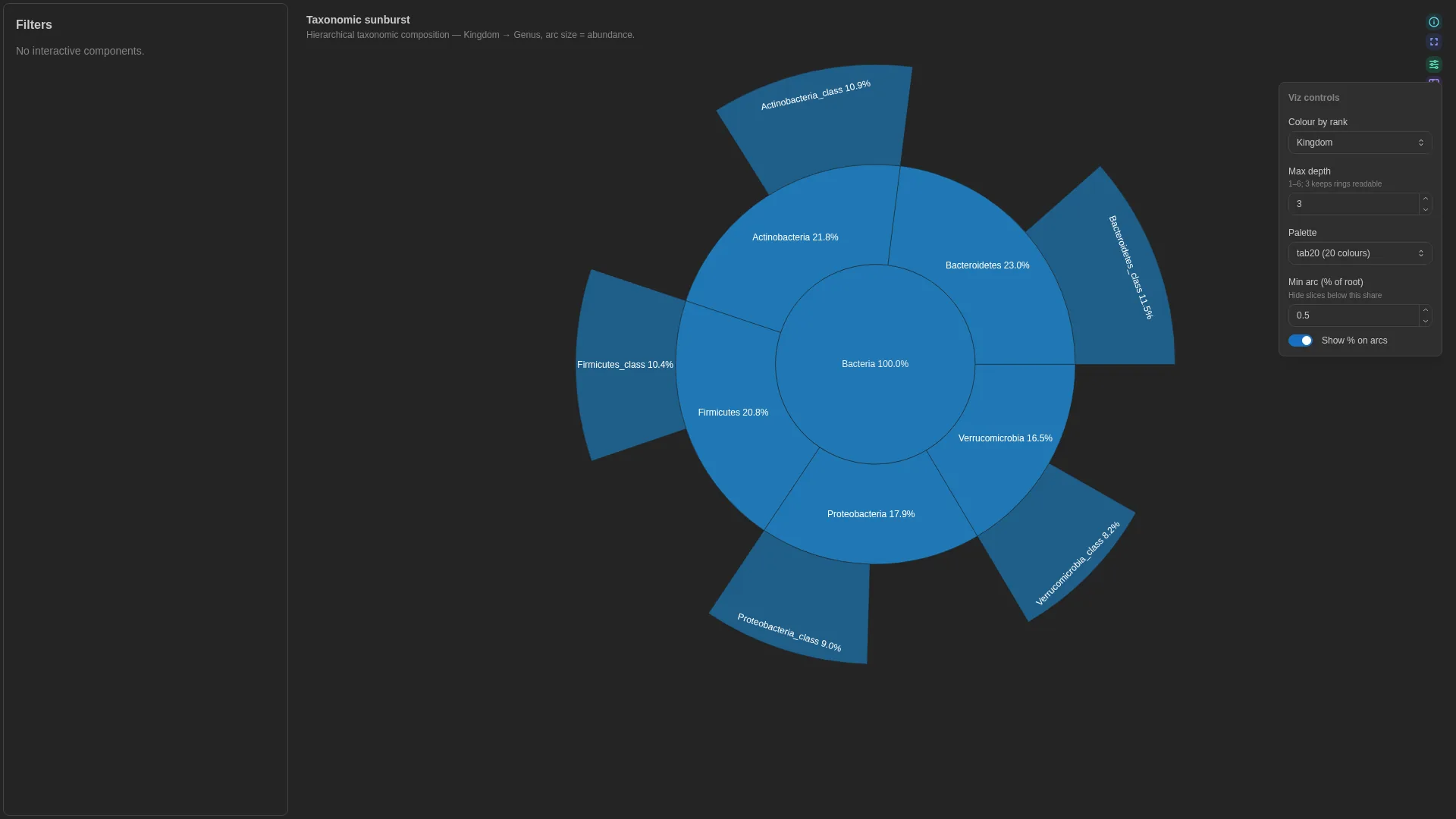
Sunburst¶
Hierarchical taxonomy / pathway viewer — concentric rings from root to leaf. Unlike most viz, Sunburst uses a multi-column rank_cols list rather than the standard single-column <role>_col pattern; the abundance role is bound via the abundance_col setting below.
Bracken vs Kraken2 — what to ingest
A raw .bracken file is flat for a single target rank (columns: name, taxonomy_id, taxonomy_lvl, fraction_total_reads, …) and does not carry explicit Kingdom→Genus rank columns. To bind it to Sunburst, either (a) ingest the Kraken2 .kreport instead and pivot the indented lineage into rank columns, or (b) map each Bracken taxonomy_id back to the NCBI taxonomy tree (e.g. taxonkit lineage, ete3) and expand to rank columns before upload. The DC must end up with one column per rank used in rank_cols plus one numeric abundance_col.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
abundance |
✓ | Numeric | Leaf abundance weight (bound via abundance_col). For Bracken: fraction_total_reads or new_est_reads. |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
rank_cols |
list[str] (≥2) | required | Hierarchical columns from root to leaf (e.g. [Kingdom, Phylum, Class, Order, Family, Genus]) |
abundance_col |
str | required | DC column that satisfies the abundance role above |
category_palette |
dict[str, str] | null | null |
Explicit value→colour overrides for the colour-key categories (whichever rank the user's Colour-by picker chooses). Pin domain palettes (e.g. Habitat → Set1) so the same category lands on the same colour across PCoA / UpSet / heatmap tiles. |
Filtering / row tagging
Renderer hierarchically aggregates by the rank_cols sequence. Intermediate arc sizes are reconstructed via Plotly's branchvalues='total'. No per-row tag — aggregation is deterministic and lossless.
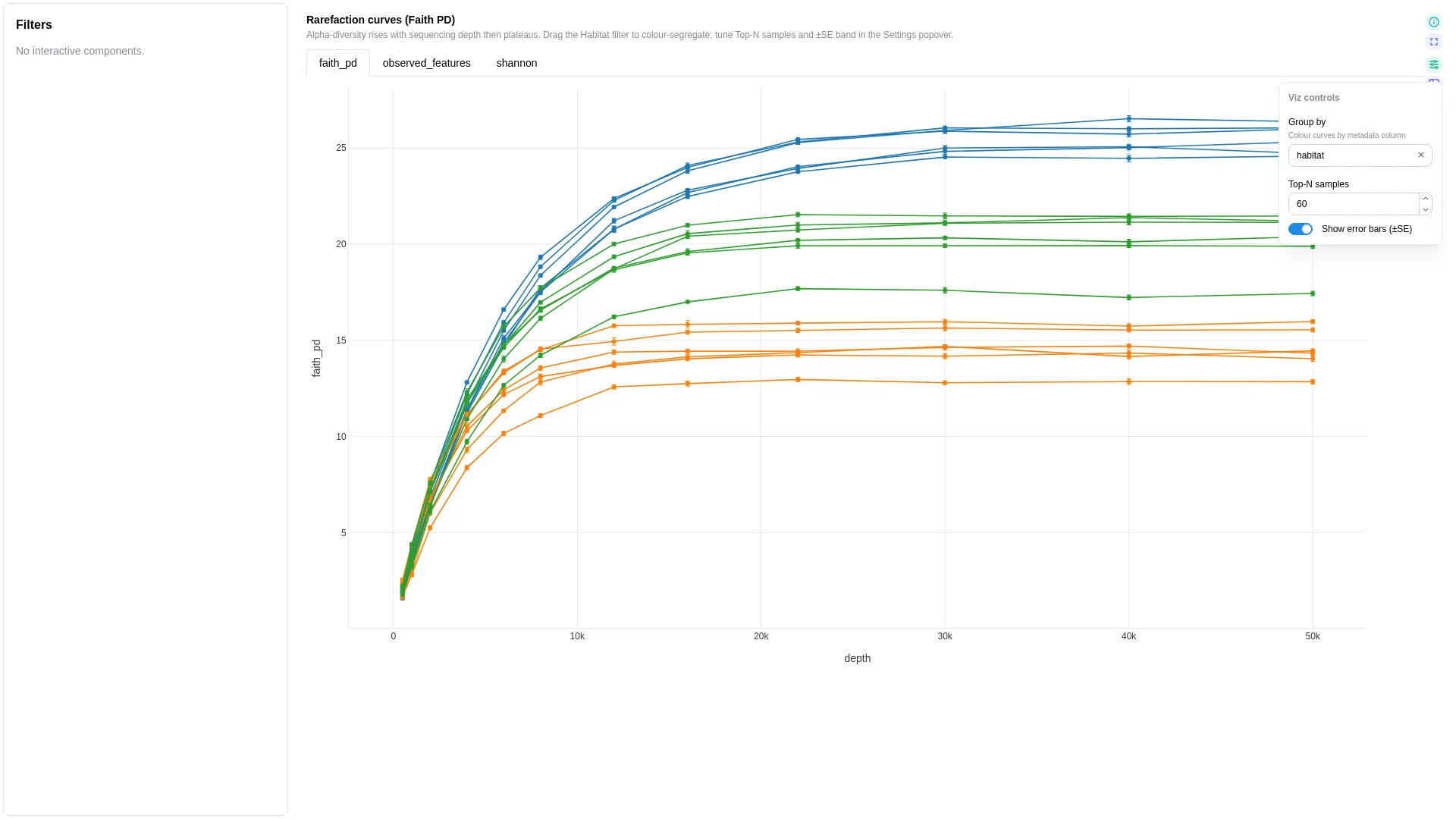
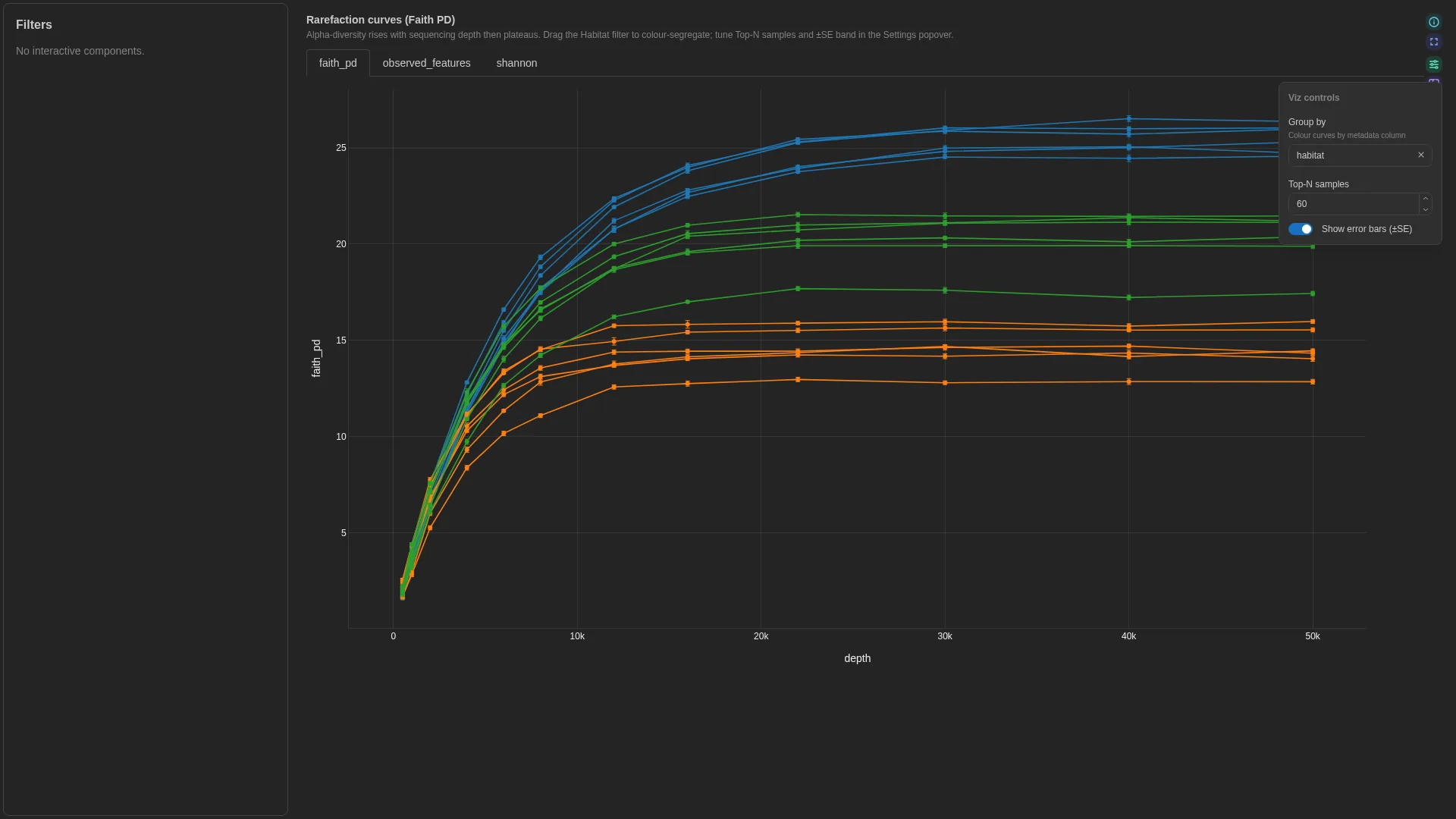
Rarefaction¶
Alpha-diversity vs sequencing depth — one line per sample with optional ±SE band and group colouring.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
sample_id |
✓ | String | Sample identifier |
depth |
✓ | Numeric | Subsampling depth (x-axis) |
metric |
✓ | Numeric | Alpha-diversity metric value (y-axis) |
iter |
— | Numeric | Iteration column to aggregate over |
group |
— | String | Categorical column for line colour grouping |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
show_ci |
bool | true |
Shade ±1 SE band around each sample's curve |
category_palette |
dict[str, str] | null | null |
Explicit value→colour overrides for the group categories. Pins domain palettes (e.g. habitat → Set1) across PCoA + UpSet + heatmap + rarefaction for cross-tab consistency. |
Filtering / row tagging
Renderer aggregates over iter per (sample_id, depth) — computes mean ± CI. Optional group adds a colour split; otherwise one line per sample. No binary row tag.
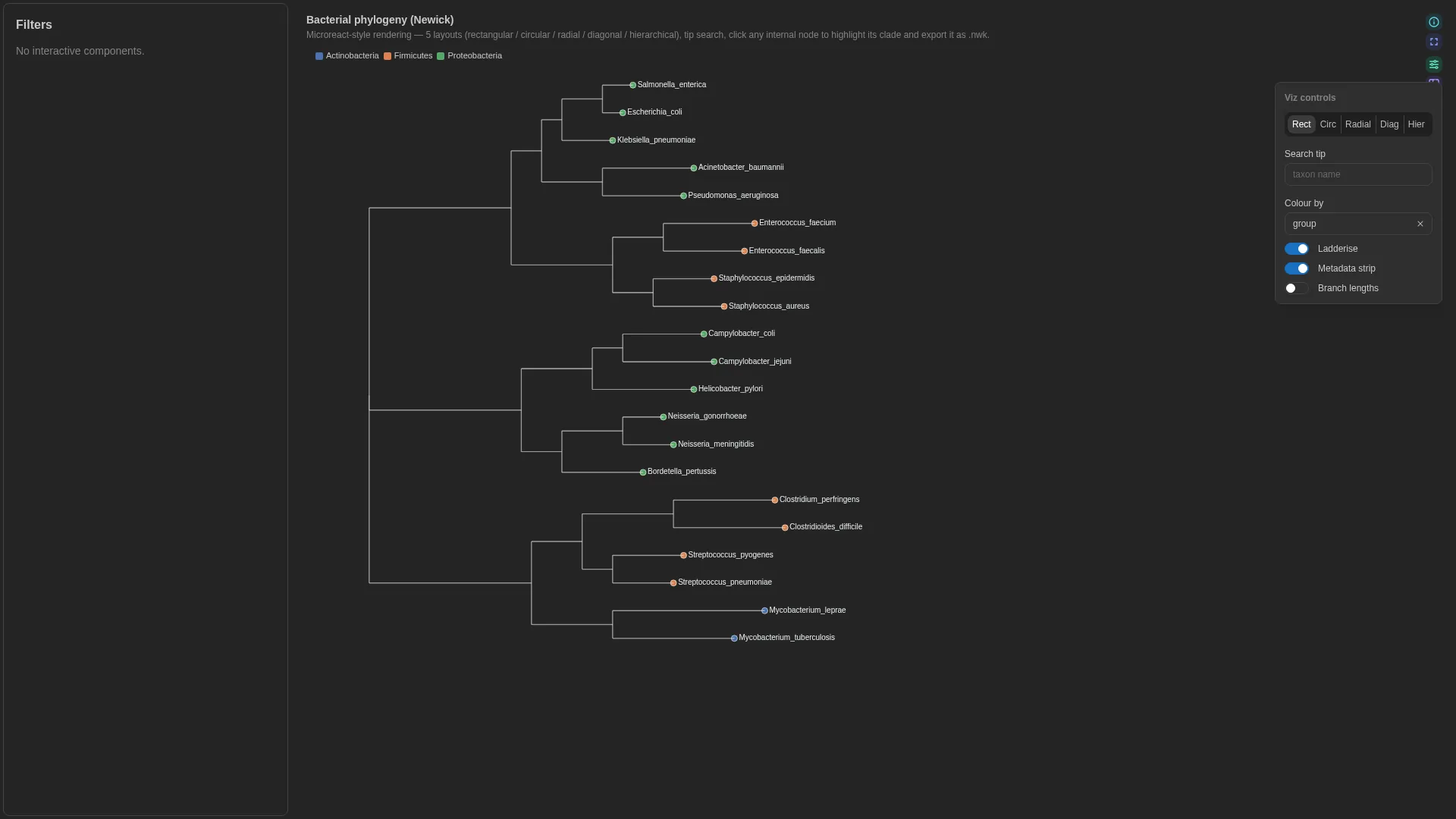
Phylogenetic¶
Newick tree + tip metadata (Microreact-style) — 5 layouts, tip search, subtree highlight.
The tree itself comes from a separate DC with dc_type: phylogeny (served via /advanced_viz/phylogeny/{dc_id}/newick). Tip annotations live in a regular Table DC and are joined to tip labels at render time via taxon_col. The schema below validates the metadata DC only.
Columns (metadata DC)
| Role | Required | Type | Description |
|---|---|---|---|
taxon |
✓ | String | Joins metadata rows to tip labels in the tree |
color |
— | Numeric | String | Tip colouring (categorical or continuous) |
label |
— | String | Metadata column shown alongside the tip label (e.g. clade name) |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
tree_wf_id / tree_dc_id |
str | required | Workflow + DC ids of the phylogeny DC |
metadata_wf_id / metadata_dc_id |
str | null | null |
Optional metadata DC for tip annotations |
taxon_col |
str | "taxon" |
Column in the metadata DC matching tip labels in the tree |
color_col |
str | null | null |
Metadata column for tip colouring (categorical or continuous) |
label_col |
str | null | null |
Metadata column shown alongside the tip label (e.g. clade name) |
extra_color_cols |
list[str] | null | null |
Extra metadata columns to pre-fetch so they appear in the viz Colour-by Select. Typical use: taxonomic ranks on ASV trees (Kingdom/Phylum/.../Species) so the user can re-colour tips at a different rank without reloading. |
category_palettes |
dict[str, dict[str, str]] | null | null |
Per-column palette overrides for the Colour-by selector. Shape: {column_name: {category_value: hex}}. Pin domain palettes (e.g. dominant_habitat → Set1) so the same category lands on the same colour across PCoA / UpSet / heatmap / phylogeny tiles. |
default_layout |
rectangular | circular | radial | diagonal | hierarchical |
rectangular |
Initial tree layout |
ladderize |
bool | true |
Ladderise the tree by default |
show_metadata_strip |
bool | true |
Render Microreact-style metadata strip next to each tip |
show_branch_lengths |
bool | true |
Annotate branches with lengths |
show_internal_labels |
bool | false |
Annotate internal nodes with their labels |

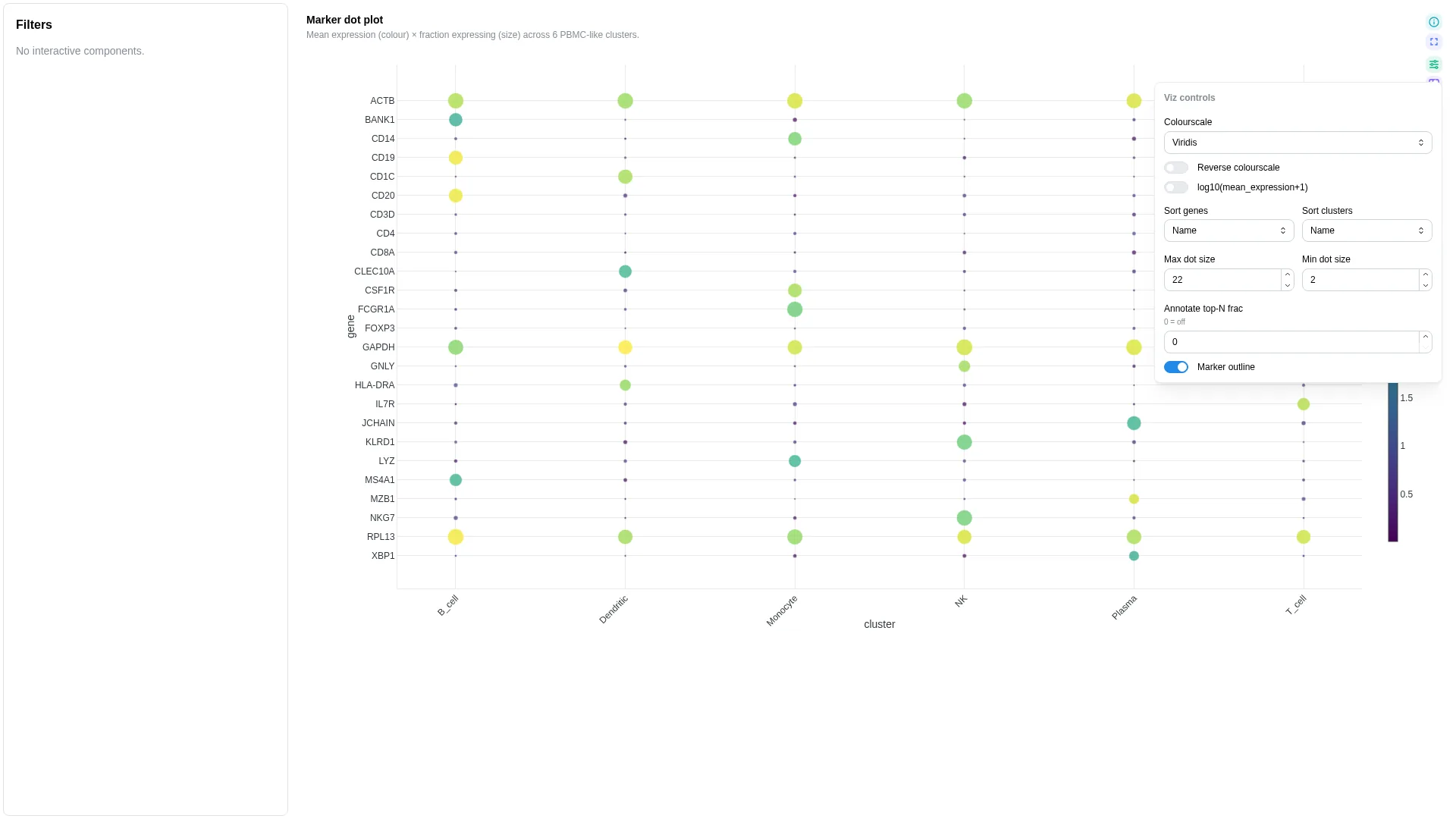
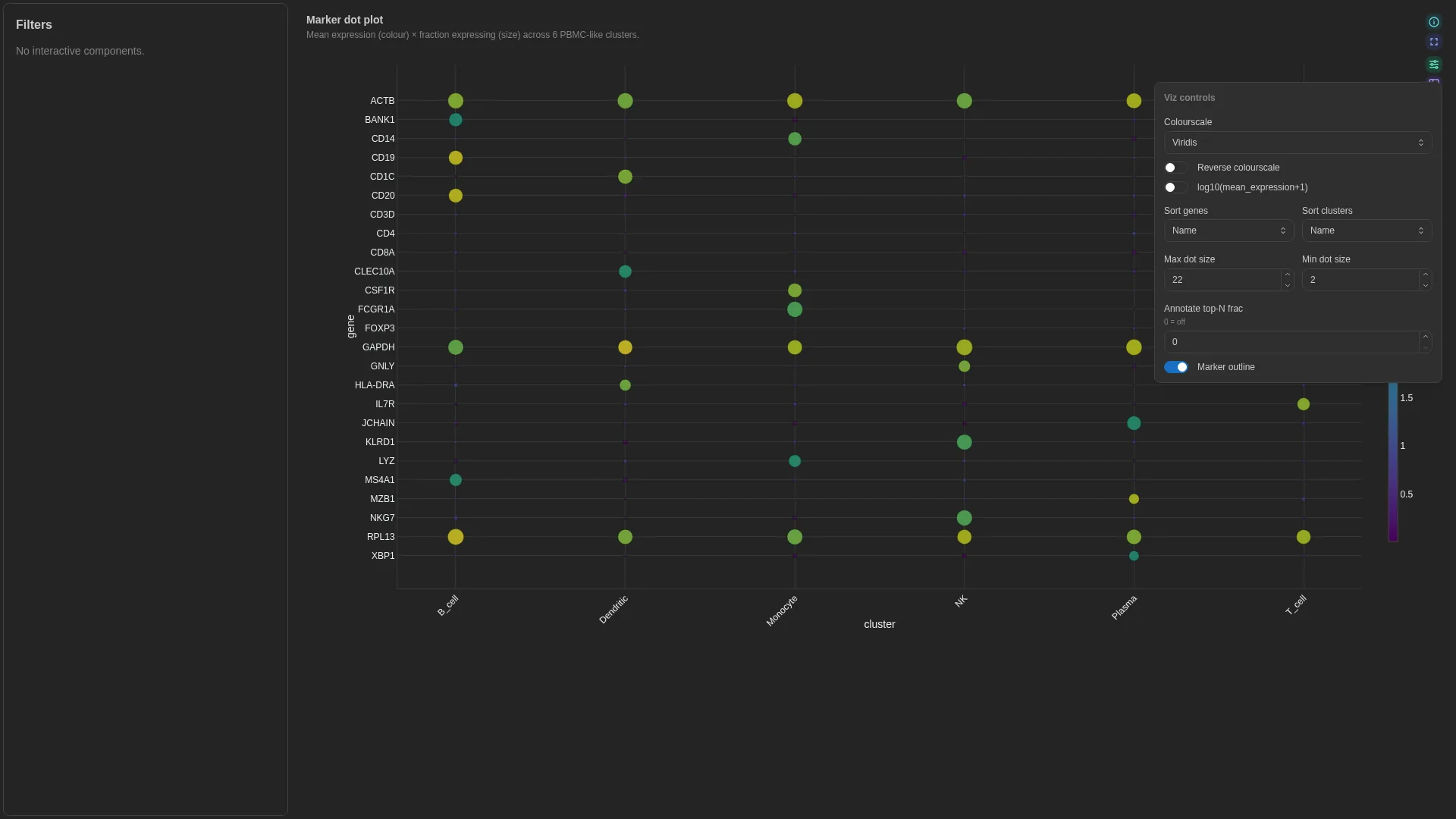
Dot plot¶
scanpy / Seurat marker-gene dot plot — cluster × gene with size = fraction expressing, colour = mean expression. The schema expects a cluster-aggregated long table — derive it from AnnData via sc.get.aggregate (or a manual groupby on adata.X), not from rank_genes_groups (which returns DE statistics, not aggregates).
Columns
| Role | Required | Type | Description |
|---|---|---|---|
cluster |
✓ | String | Cluster / group (x-axis) |
gene |
✓ | String | Gene / feature (y-axis) |
mean_expression |
✓ | Float | Mean expression per (cluster, gene) — dot colour |
frac_expressing |
✓ | Float | Fraction of cells with X > 0 per (cluster, gene) — dot size |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
max_dot_size |
int (4–60) | 22 |
Max marker size in pixels |
min_dot_size |
int (0–20) | 2 |
Min marker size in pixels |
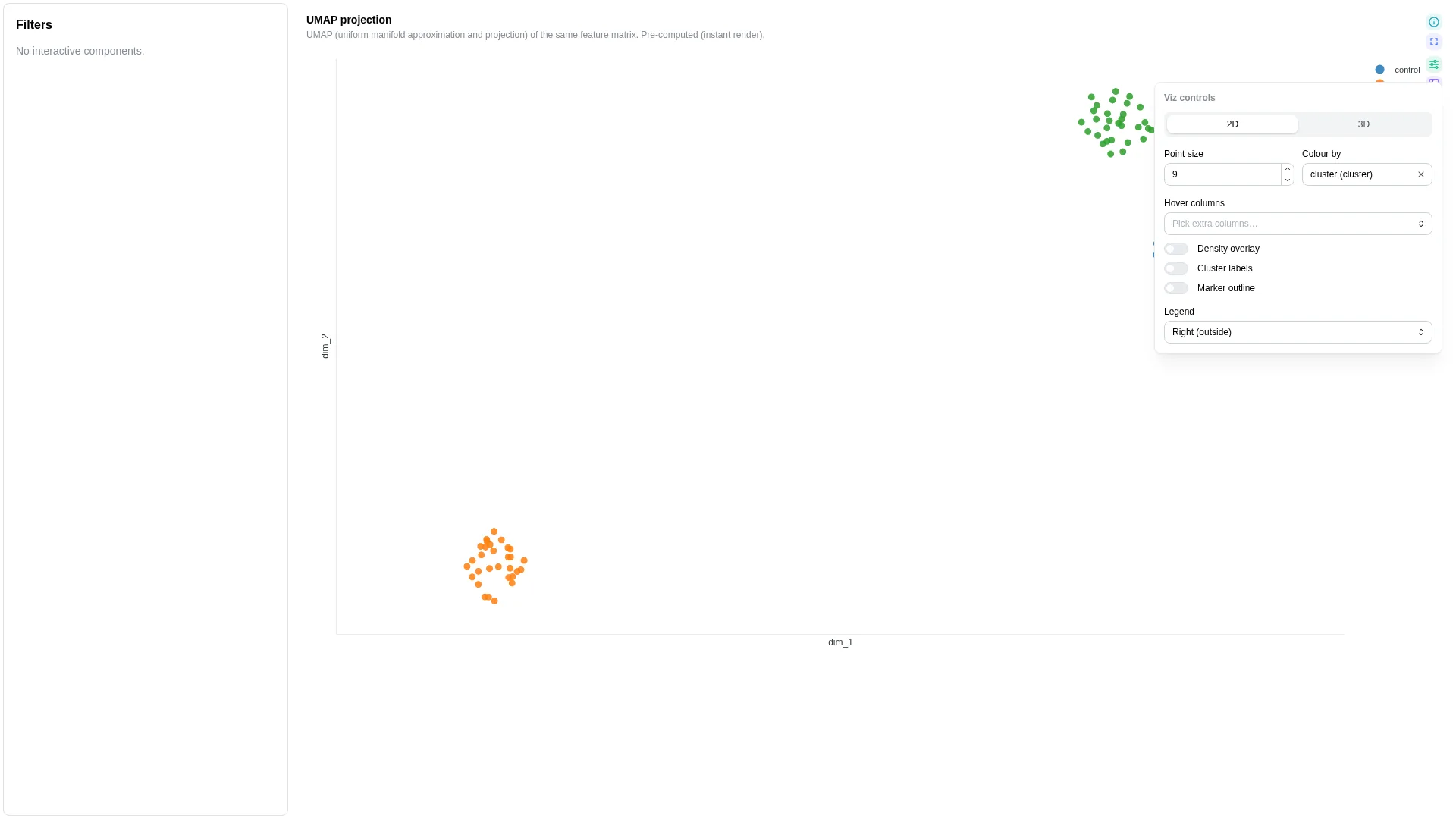
Embedding¶
2D / 3D sample embedding (PCA / UMAP / t-SNE / PCoA) — supports a precomputed DC (dim_1, dim_2 columns already materialised) or live-compute mode (run the reduction on the fly via a Celery task and cache by (dc, method, params, filters)).
Columns
| Role | Required | Type | Description |
|---|---|---|---|
sample_id |
✓ | String | Sample identifier |
dim_1 |
✓ | Float | First embedding dim (precomputed mode) |
dim_2 |
✓ | Float | Second embedding dim (precomputed mode) |
dim_3 |
— | Float | Third dim — enables 3D |
cluster |
— | String | Cluster assignment column |
color |
— | Numeric | String | Point colouring (metadata or expression) |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
compute_method |
pca | umap | tsne | pcoa | null |
null |
When set, runs the reduction live on the server; null = precomputed mode |
umap_n_neighbors |
int (2–100) | 15 |
UMAP n_neighbors |
umap_min_dist |
float (0–1) | 0.1 |
UMAP min_dist |
tsne_perplexity |
float (2–100) | 30.0 |
t-SNE perplexity |
tsne_n_iter |
int (250–5000) | 1000 |
t-SNE iterations |
pcoa_distance |
bray_curtis |
bray_curtis |
PCoA distance metric |
show_density |
bool | false |
Overlay density contours |
point_size |
int (1–30) | 6 |
Marker size |
category_palette |
dict[str, str] | null | null |
Explicit value→colour overrides for the categorical color column. Wins over the default palette-index assignment so dashboards can pin domain-specific colours (e.g. habitat → Set1) without forking the renderer per project. |
Filtering / row tagging
In precomputed mode the renderer just plots the pre-existing coordinates. In live-compute mode it dispatches POST /advanced_viz/compute_embedding, which runs the chosen reduction on the wide sample × feature matrix and returns coordinates. Results are cached by (dc_id, method, params, filters); tweaking a slider re-dispatches a fresh job.
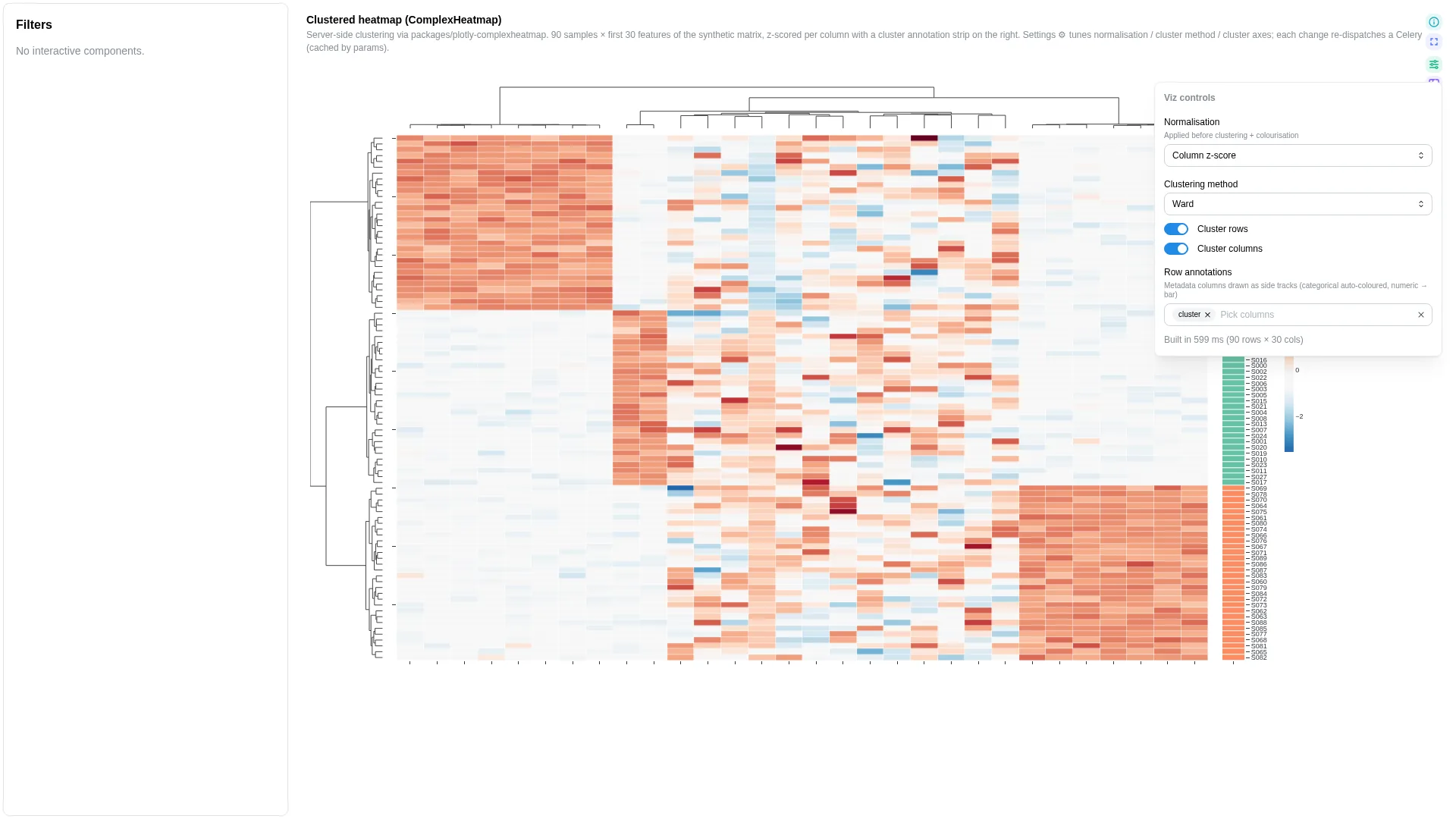
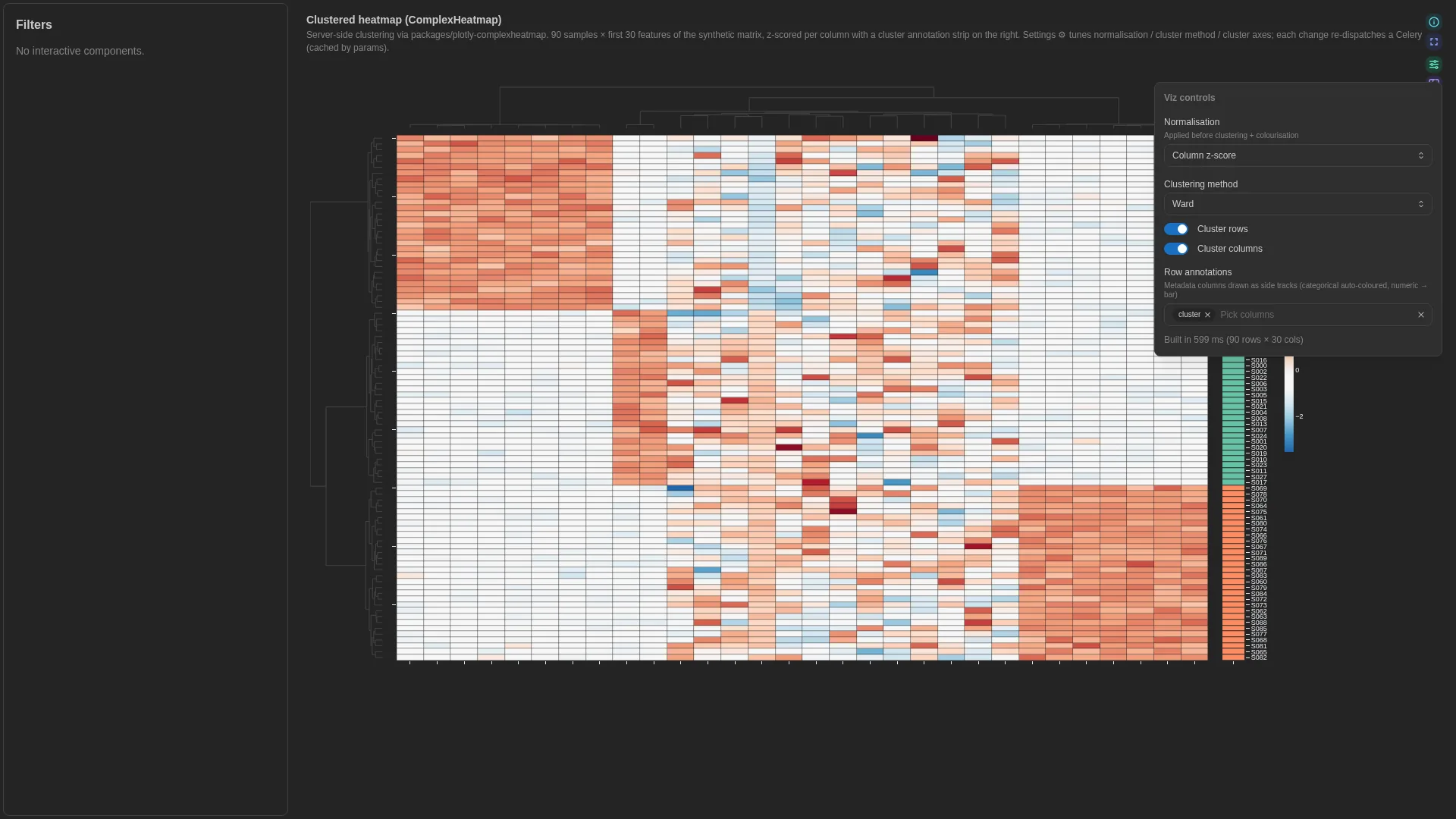
Hierarchical Heatmap¶
Clustered heatmap with dendrograms + annotation tracks, à la R's ComplexHeatmap / pheatmap. Wraps the in-tree plotly-complexheatmap library; heavy compute (clustering, dendrogram layout) runs in a Celery worker and is cached by (dc, params hash).
Columns
| Role | Required | Type | Description |
|---|---|---|---|
index |
✓ | String | Row-label column (typically sample_id) |
Numeric matrix columns are inferred from the rest of the DC schema at compute time — there's no per-role binding for the value columns.
Settings
| Option | Type | Default | Description |
|---|---|---|---|
matrix_wf_id / matrix_dc_id |
str | required | Workflow + DC ids of the wide matrix DC |
index_column |
str | sample_id |
Row-label column |
value_columns |
list[str] | null | null |
Subset of numeric columns; null = all numeric |
row_annotation_cols |
list[str] | [] |
Categorical columns rendered as a right-side annotation strip |
col_annotations |
dict[str, dict[str, str]] | null | null |
Per-column categorical annotations rendered as a top strip. Shape: {annotation_name: {column_label: category_value}} (e.g. {'habitat': {'SRR10070130': 'Riverwater', ...}}). The renderer aligns the values to the matrix's column order. Use when per-sample metadata (treatment / habitat / batch) needs to live on the column axis without joining a second DC. |
col_annotation_colors |
dict[str, dict[str, str]] | null | null |
Per-annotation palette overrides for the column-annotation track. Shape: {annotation_name: {category_value: hex}}. When unset the server picks colours from a Dark2 palette (chosen to contrast with the row-track's Set2 pastels). Use to pin domain palettes (e.g. habitat → Set1) across PCoA + UpSet + heatmap. |
cluster_rows / cluster_cols |
bool | true |
Enable hierarchical clustering |
cluster_method |
ward | single | complete | average |
ward |
Linkage method |
cluster_metric |
euclidean | correlation | cosine |
euclidean |
Distance metric |
normalize |
none | row_z | col_z | log1p |
none |
Pre-clustering normalisation |
colorscale |
str | null | null |
Plotly colorscale name override |
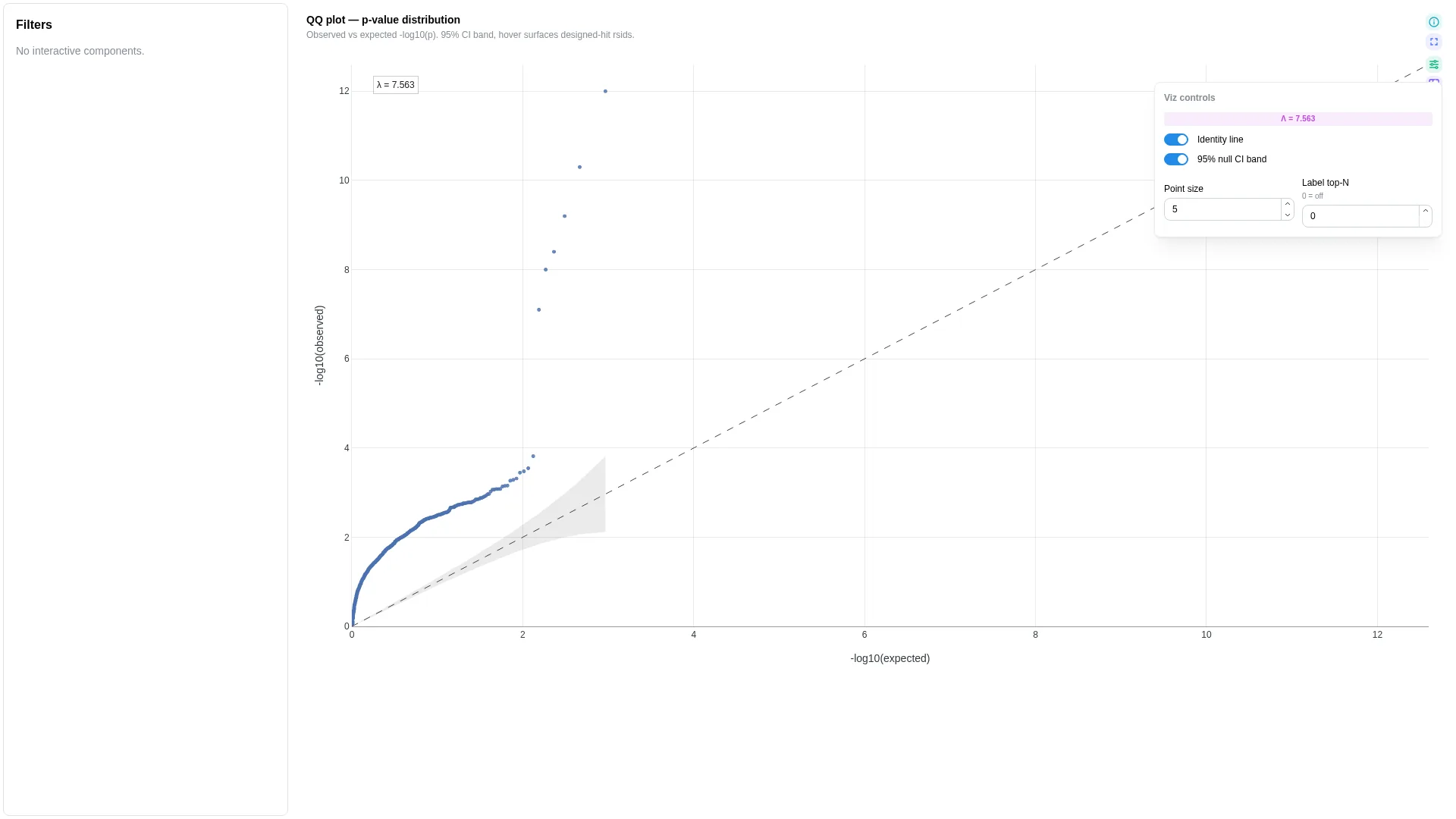
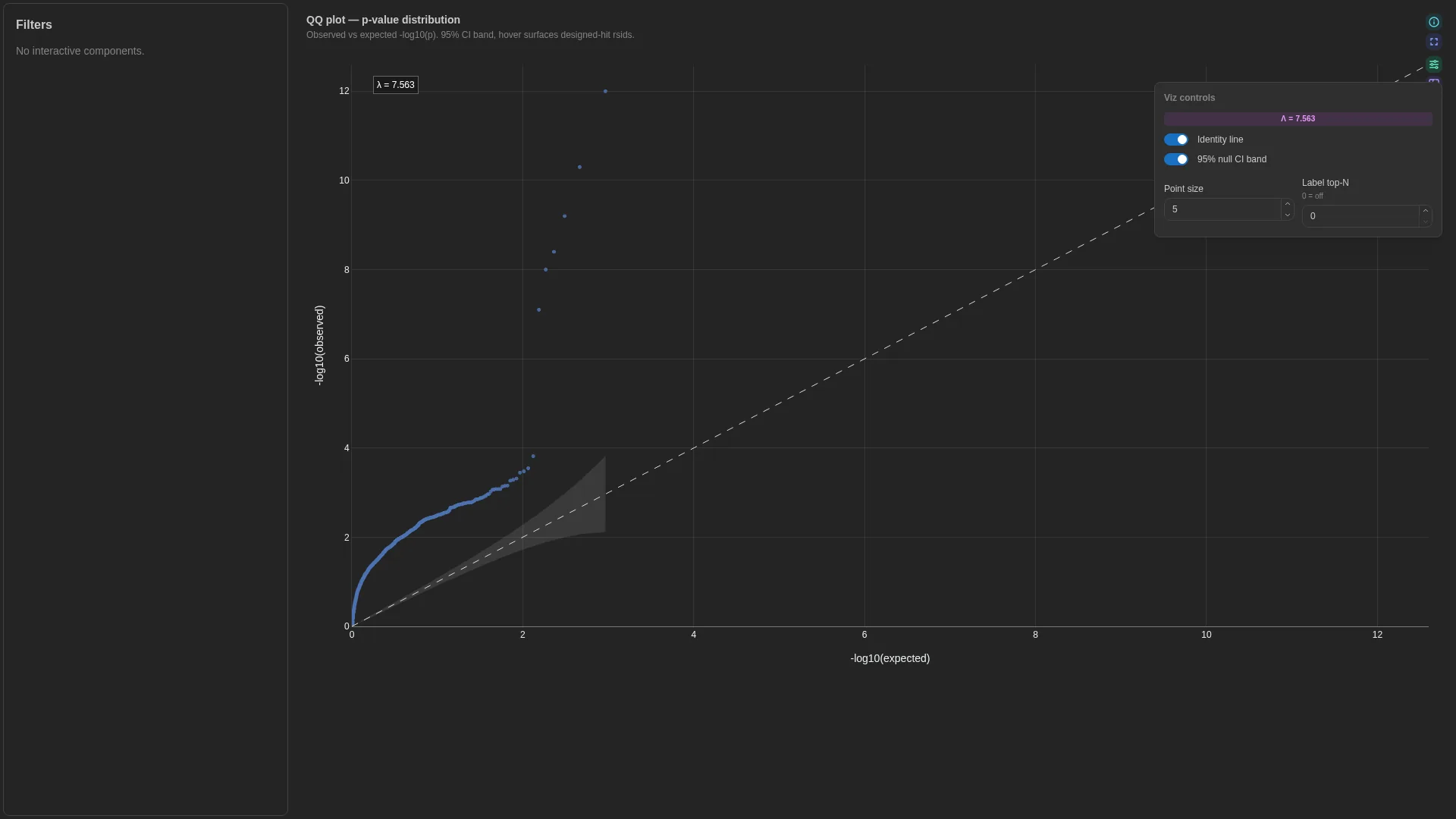
QQ¶
Quantile-quantile plot for p-value distributions (GWAS / DE / eQTL QC). Sorts p-values and plots -log10(observed) against the theoretical -log10(expected) under a uniform null.
Columns
| Role | Required | Type | Description |
|---|---|---|---|
p_value |
✓ | Float | Raw p-value (0–1) |
feature_id |
— | String | Hover-only id |
category |
— | String | Stratification column (one trace per stratum) |
Settings
| Option | Type | Default | Description |
|---|---|---|---|
show_ci |
bool | true |
Shade the 95% null CI band |
Filtering / row tagging
When category is bound, the renderer produces one trace per stratum (e.g. genome partitions, ancestry groups). Otherwise a single trace plus the y = x reference line and the optional 95% null CI band.
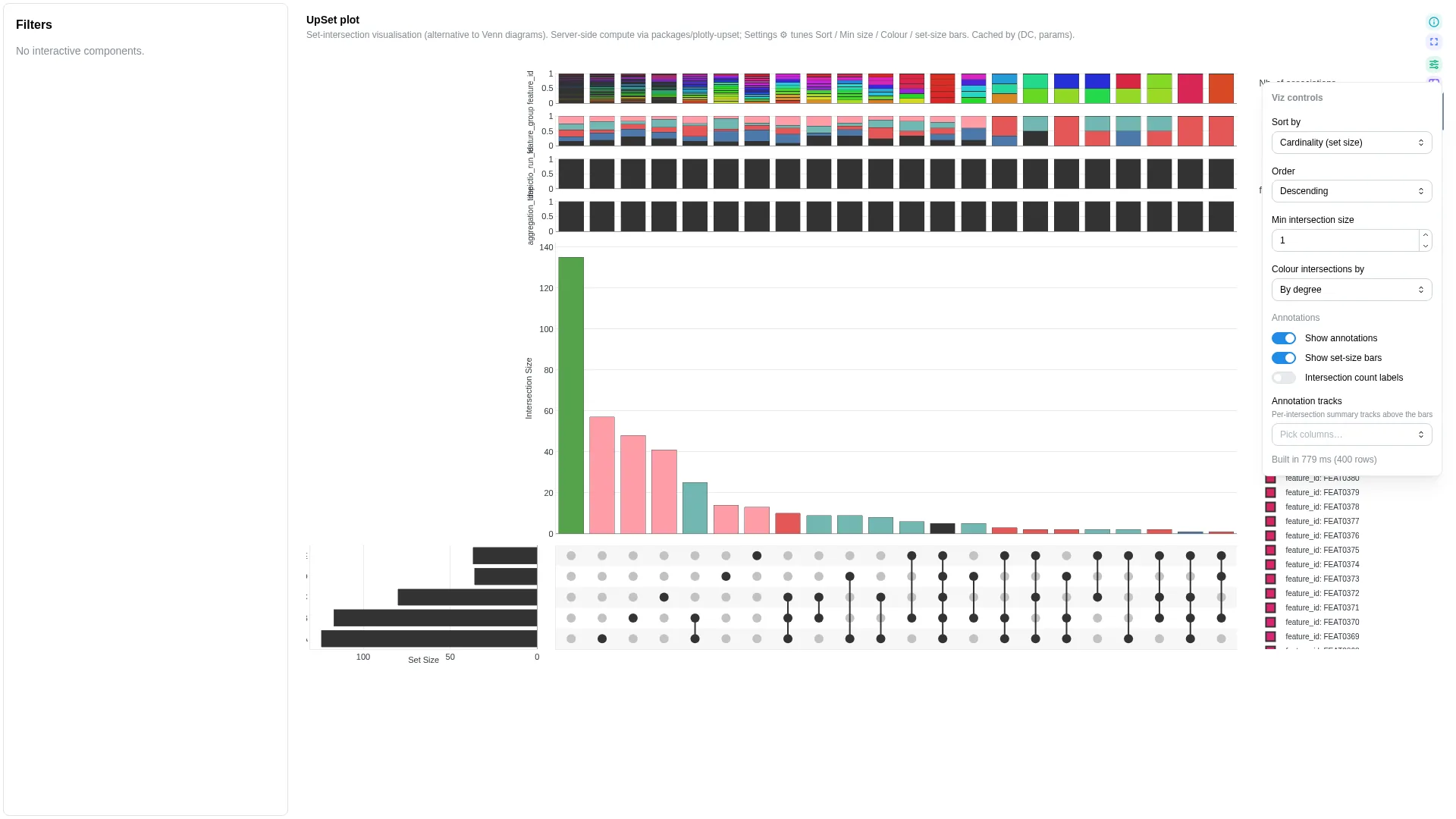
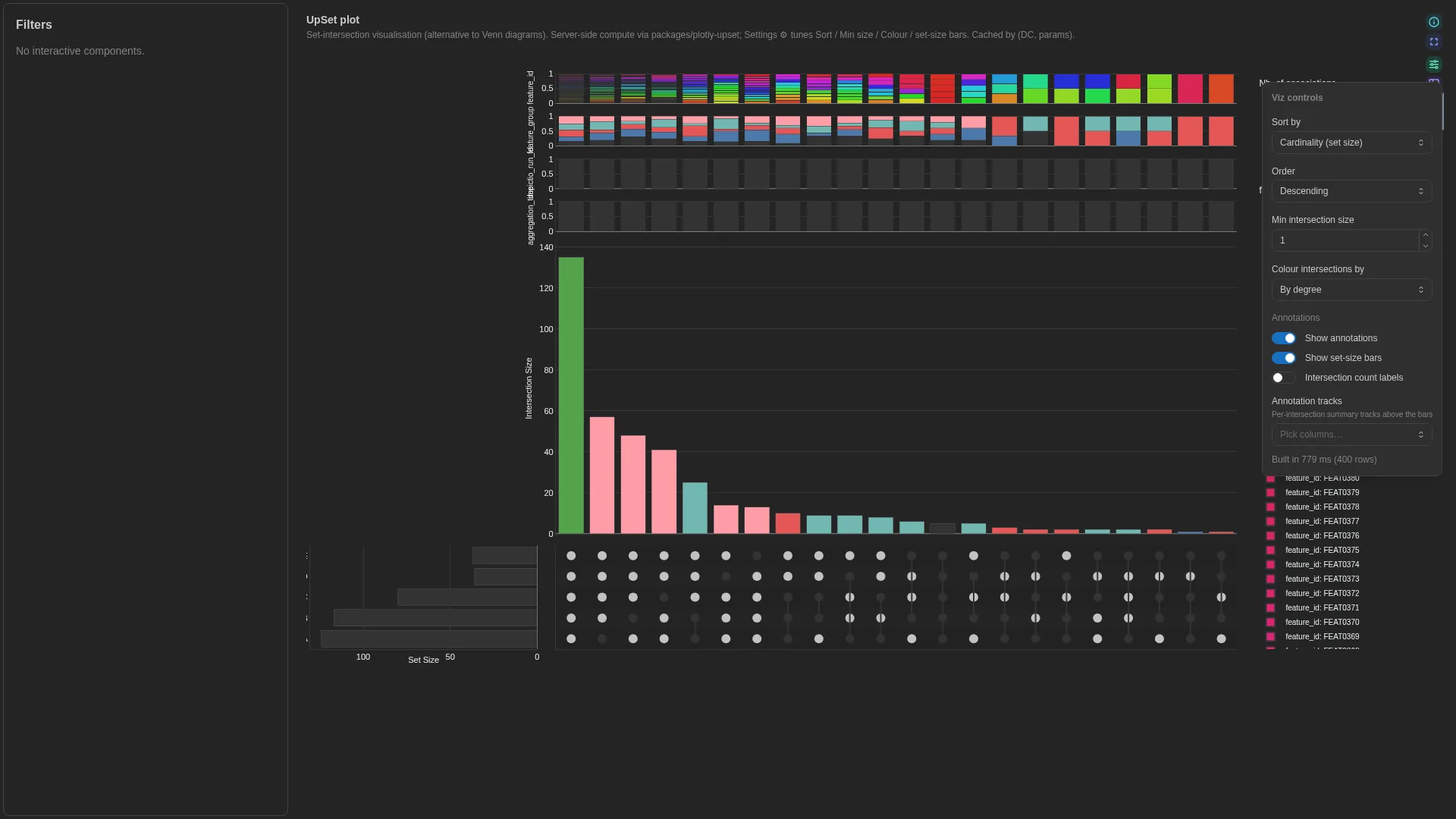
UpSet¶
Set-intersection visualisation (alternative to Venn diagrams). Wraps the in-tree plotly-upset library (vendored under packages/plotly-upset/, not yet a standalone public repo); intersection enumeration + sorting runs in a Celery worker and is cached by (dc, params hash). Input DC: a binary table where each row is an element and each set_col is a 0/1 membership indicator.
Columns
No canonical role-based schema — the renderer enumerates binary columns at compute time. Editor binding validation is a no-op.
Settings
| Option | Type | Default | Description |
|---|---|---|---|
matrix_wf_id / matrix_dc_id |
str | required | Workflow + DC ids of the membership DC |
set_columns |
list[str] | null | null |
Explicit list of set columns; null = auto-detect binary |
sort_by |
cardinality | degree | degree-cardinality | input |
cardinality |
Intersection ordering |
sort_order |
descending | ascending |
descending |
Ordering direction |
min_size |
int (≥0) | 1 |
Hide intersections smaller than this |
max_degree |
int | null | null |
Hide intersections involving more than N sets |
show_set_sizes |
bool | true |
Show horizontal set-size bar chart |
color_intersections_by |
none | set | degree |
none |
Intersection-bar colour mode |
set_colors |
dict[str, str] | null | null |
Per-set colour overrides (set name → hex). Drives set-size bars + matrix dots + intersection bars (when color_intersections_by="set"). Pin domain palettes (e.g. habitat → Set1) so the same set lands on the same colour across tiles. |
Filtering / row tagging
Renderer filters by intersection size and degree — min_size drops intersections below the threshold, max_degree drops intersections involving more sets than the limit.
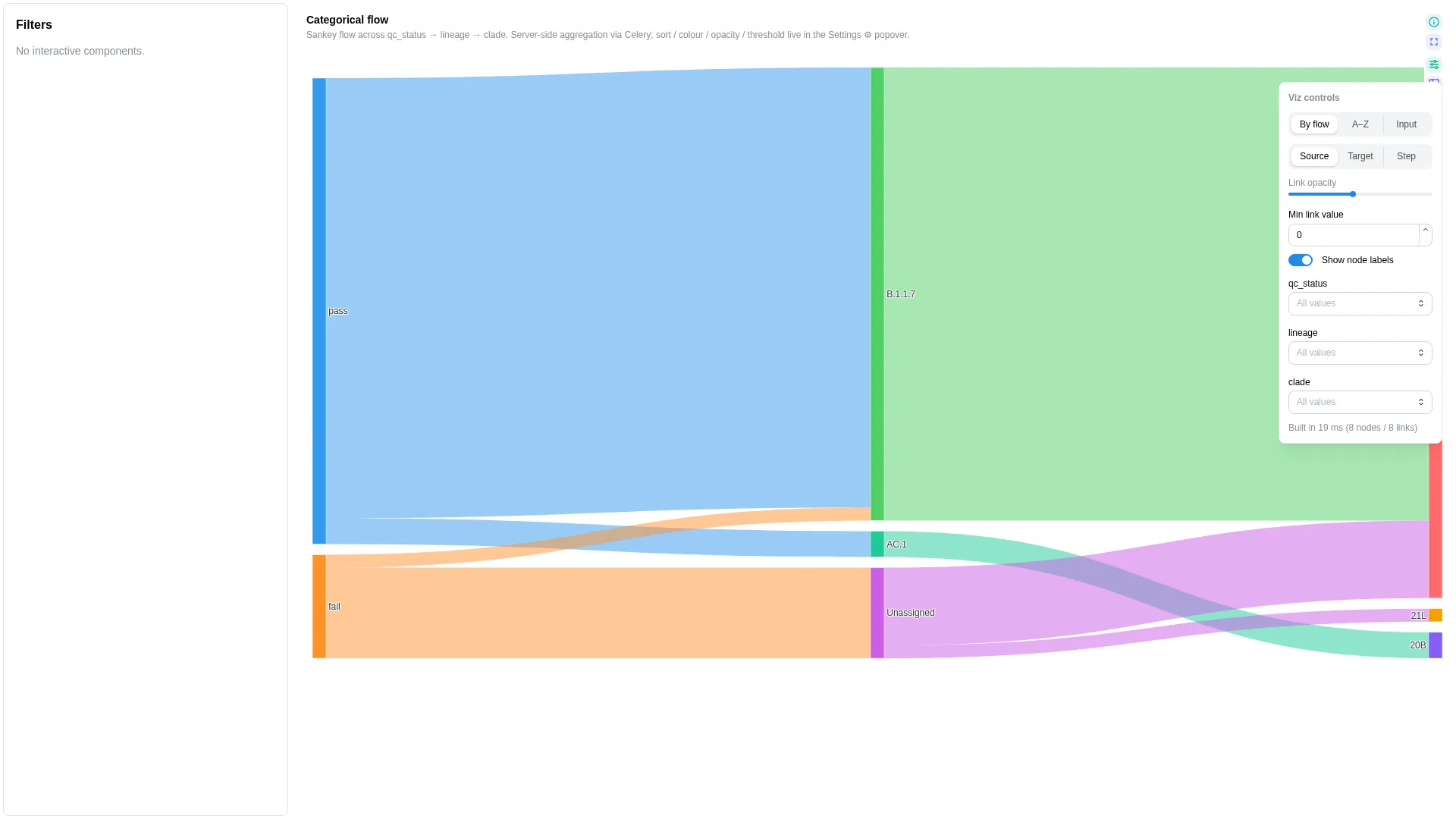
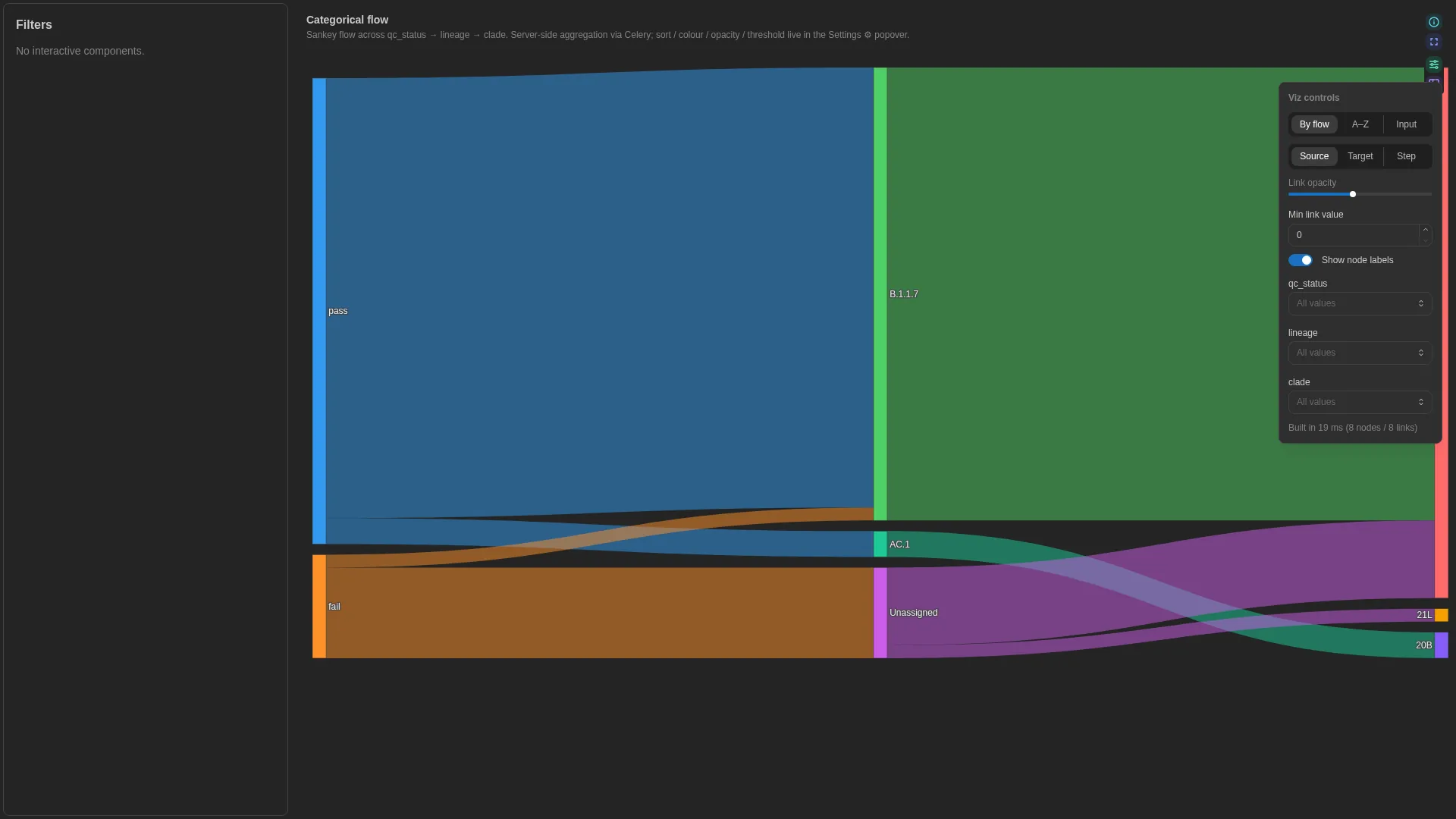
Sankey¶
Categorical-flow diagram across N ordered categorical levels (e.g. sample → lineage → clade, sample → kingdom → phylum → genus). Server-side aggregation via Celery, client-side colour / opacity tweaks.
Columns
No canonical role-based schema — step_cols is a multi-column list (≥2 ordered categorical columns). The renderer validates step presence at compute time; the editor enforces min_length=2.
Settings
| Option | Type | Default | Description |
|---|---|---|---|
step_cols |
list[str] (≥2, unique) | required | Ordered categorical columns from source to leaf |
available_step_cols |
list[str] | null | null |
Full ordered list of columns the user can wire as steps. When set, the renderer exposes a Depth slider that picks the first N columns from this list; step_cols becomes the initial prefix. Leaving it null locks the diagram to step_cols. |
value_col |
str | null | null |
Optional numeric weight; null = each row counts as 1 |
value_label |
str | null | null |
Human-readable label for value_col shown in hover tooltips. Defaults to value_col when unset (e.g. "abundance"). |
value_format |
raw | fraction | count |
raw |
Hover display mode. fraction multiplies by 100 and appends %; count uses thousands separators; raw adapts decimal precision to magnitude. |
sort_mode |
alphabetical | total_flow | input |
total_flow |
Node-ordering rule |
color_mode |
source | target | step |
source |
Link colouring rule |
link_opacity |
float (0.05–1) | 0.5 |
Link transparency |
min_link_value |
float (≥0) | 0.0 |
Hide links whose aggregated value is below this threshold |
show_node_labels |
bool | true |
Render node labels |
Recipe-coupled normalisation (ampliseq)
The bundled sankey_canonical recipe for nf-core/ampliseq pre-divides per-sample relative abundance by sample count so the renderer's sum-aggregation reads as a mean-per-sample at the root (≈1.0 = 100%). This bakes a sum-aggregation assumption into the canonical DC and is recomputed once at recipe time — cross-DC sample filters do not rescale the divisor, so heavy filtering yields scaled-down totals. Treat the values as relative composition, not absolute. See depictio/projects/nf-core/ampliseq/recipes/sankey_canonical.py for the full caveat.
Filtering / row tagging
Renderer aggregates by the step_cols sequence (via Celery compute_sankey) and filters out links whose aggregated value is below min_link_value.
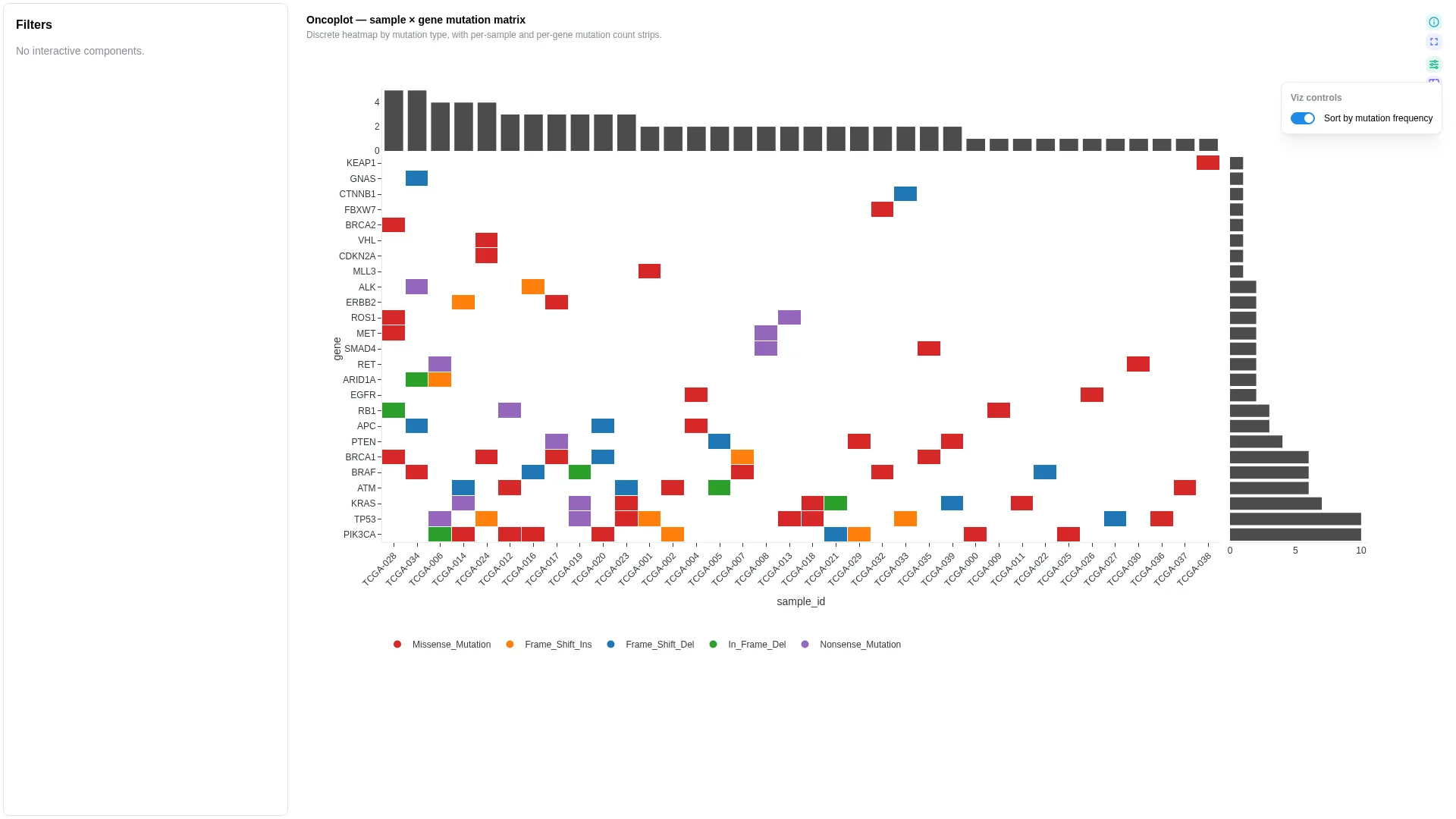
Oncoplot¶
Sample × gene mutation matrix with discrete mutation-type colours and per-gene / per-sample frequency strips.
Columns — straight rename from canonical Mutation Annotation Format (VEP / vcf2maf / maftools — the cancer-mutation file format, not Minor Allele Frequency):
| Role | Required | Type | Description | Mutation Annotation Format column |
|---|---|---|---|---|
sample_id |
✓ | String | Sample identifier (x-axis) | Tumor_Sample_Barcode |
gene |
✓ | String | Gene identifier (y-axis) | Hugo_Symbol |
mutation_type |
✓ | String | Categorical mutation type (cell colour) | Variant_Classification |
Settings
No additional knobs — the layout is fully determined by the column bindings.
Filtering / row tagging
Cells are coloured categorically by mutation_type (NA cells stay blank). Side strips show per-gene and per-sample mutation counts.
Card Components (v0.0.1+)¶
Card components display metrics with aggregations. A card shows a hero metric (primary value) and can optionally display secondary metrics below it for at-a-glance summaries.
Aggregation Types¶
| Aggregation | Description | Example |
|---|---|---|
| count | Number of rows | Total samples |
| sum | Sum of values | Total reads |
| average | Average value | Average coverage |
| median | Median value | Median quality score |
| min | Minimum value | Minimum mapping rate |
| max | Maximum value | Maximum duplication rate |
| nunique | Count of unique values | Unique sample types |
| std_dev | Standard deviation | Coverage spread |
| variance | Variance | Expression variability |
| range | Max − Min | Read length range |
| skewness | Distribution skew | Quality score symmetry |
| kurtosis | Distribution tailedness | Outlier tendency |
| percentile | 50th percentile | Median coverage |
| mode | Most frequent value | Dominant sample type |
| q1 | 25th percentile | Lower-quartile coverage |
| q3 | 75th percentile | Upper-quartile coverage |
| box_plot_stats | Tukey 5-number summary (min, Q1, median, Q3, max) | Required by secondary_layout: box_plot |
Configuration¶
- Select a Data Collection
- Choose a Column to aggregate
- Select an Aggregation type (hero metric)
- Optionally add Secondary Metrics (
aggregations) for a multi-metric summary - Optionally add a Filter Expression (
filter_expr) for conditional aggregation - Customize the Title, Icon, and Styling
Multi-Metric Summary Cards¶
Cards can display multiple aggregation results in a single component. The primary aggregation is shown as the large hero value, and aggregations are displayed as compact secondary rows below it.
┌─────────────────────────────────┐
│ Petal Length [Icon]│
├─────────────────────────────────┤
│ 4.5 cm │
│ (Average) │
├─────────────────────────────────┤
│ Median: 4.35 │
│ Std Dev: 0.82 │
│ Min: 1.00 │
│ Max: 6.90 │
└─────────────────────────────────┘
Secondary Layout Modes (v0.13.0+)¶
The default layout above stacks aggregations as a vertical list under the hero metric. Set secondary_layout on the card to switch to one of five richer layouts — each tuned for a specific summary intent and with its own required companion field.
secondary_layout |
Renders | Companion fields required |
|---|---|---|
vertical (default) |
Stacked rows from aggregations list |
aggregations |
compact |
Horizontal strip from aggregations |
aggregations |
box_plot |
Tukey box-and-whisker (min / Q1 / median / Q3 / max) | aggregations: [box_plot_stats] |
top_n |
Mini bar chart of top-N most frequent breakdown_col values |
breakdown_col, top_n_count (1–5) |
coverage |
Fill bar showing value / coverage_max |
coverage_max |
concentration |
Top-N share (%) by breakdown_col |
breakdown_col, top_n_count (1–5) |
YAML field reference (multi-metric extras)
| Field | Type | Default | Description |
|---|---|---|---|
secondary_layout |
enum (see table above) | vertical |
Layout mode for the secondary block. |
aggregations |
list[str] | null | null |
Secondary aggregation functions (e.g. [median, std_dev, min, max] or [box_plot_stats]). Required by vertical, compact, box_plot. |
breakdown_col |
str | null | null |
Group-by column for top_n / concentration layouts. |
top_n_count |
int (1–5) | 3 |
Number of top values rendered in top_n / concentration layouts. |
coverage_max |
float | null | null |
Denominator for the coverage layout's fill bar. Falls back to vertical if missing. |
Examples (from depictio/projects/init/iris/dashboards/overview.yaml)
Conditional Aggregation (filter_expr)¶
Cards support a filter_expr field — a Polars expression that pre-filters the data before computing the aggregation. This enables conditional metrics like "count of samples with coverage > 30x" without creating a separate data collection.
filter_expr works on top of interactive filters (dual-layer filtering): interactive filters narrow the dataset first, then filter_expr applies an additional condition before aggregation.
See Filter Expressions for the complete expression reference.
Styling Options¶
| Option | Description | Example |
|---|---|---|
| Title | Metric label | "Total Samples" |
| Description | Subtitle text | "Across all batches" |
| Icon | Iconify icon name | mdi:chart-line |
| Icon Color | Icon accent color | #2196F3 |
| Title Color | Title text color | #333333 |
| Title Font Size | Title font size | sm |
| Value Font Size | Hero value font size | xl |
Text Components (v0.2.0+)¶
Text components are presentational tiles for section delimiters, narrative intros, and small inline annotations. They have no data binding — they just render a heading + optional paragraph at the position they occupy in the grid.
YAML fields¶
| Field | Type | Default | Description |
|---|---|---|---|
component_type |
"text" |
— | Discriminator |
title |
str | "" |
Heading text |
order |
int (1–6) | 1 |
Heading level — renders as <h1> through <h6> (values outside 1–6 are clamped) |
alignment |
left | center | right |
left |
Horizontal text alignment for both title and body |
body |
str | "" |
Optional paragraph rendered below the title |
Inline markdown¶
The body and title support a limited inline subset — no markdown library is loaded; rendering is done client-side in TextRenderer.tsx:
**bold**→ bold*italic*→ italic`code`→code
Block-level constructs (lists, tables, blockquotes, fenced code, links, images) are not supported. For richer narrative content, use the dashboard's notes panel.
Example (from depictio/projects/init/iris/dashboards/overview.yaml)¶
- tag: text-overview-intro
component_type: text
title: "Iris Dataset — Overview"
order: 2
alignment: left
body: "Fisher's classic 150-flower dataset across three varieties (*Setosa*, *Versicolor*, *Virginica*). Filters on the left refine every tile."
layout: {x: 0, y: 0, w: 8, h: 1}
Use Cases¶
- Dashboard section headers
- Visual organization of content
- Labeling groups of related components
- Short narrative intros above an analytical section
Interactive Components (v0.0.1+)¶
Interactive components let users filter data across the dashboard. These components affect all linked visualization components.
Component Types¶
| Component | Input Type | Best For |
|---|---|---|
| RangeSlider | Numeric range | Coverage: 0-100x |
| MultiSelect | Multiple choices | Sample types |
| DatePicker | Date range | Run dates |
| SegmentedControl | Single choice | Condition A/B |
| TextInput | Free text | Sample ID search |
RangeSlider¶
Filter data by numeric range:
| Option | Description |
|---|---|
| Column | Numeric column to filter |
| Min/Max | Range bounds |
| Step | Increment value |
| Default | Initial range values |
MultiSelect¶
Filter by selecting multiple values:
| Option | Description |
|---|---|
| Column | Categorical column to filter |
| Options | Available values (auto-populated) |
| Default | Initially selected values |
| Placeholder | Hint text when empty |
DatePicker¶
Filter by date range:
| Option | Description |
|---|---|
| Column | Date/datetime column to filter |
| Format | Date display format |
| Default | Initial date range |
SegmentedControl¶
Single-selection toggle:
| Option | Description |
|---|---|
| Column | Column to filter |
| Options | Available choices |
| Default | Initially selected option |
Scoped Filters (filter_expr)¶
Interactive components support a filter_expr field that pre-filters the underlying data before computing component options. Instead of showing all possible values, the component shows only values that exist in the filtered subset.
Example: A MultiSelect with filter_expr: "col('petal.length') > 4" only shows variety options that appear in rows where petal length exceeds 4 cm.
This is useful for:
- Cascading filters — show only relevant options based on data conditions
- Domain-specific scoping — e.g., only show taxa above an abundance threshold
- Focused analysis — restrict a slider's range to a meaningful subset
See Filter Expressions for the complete expression reference.
MultiQC Components (v0.5.0+)¶
MultiQC components display quality control reports generated by MultiQC.
Features¶
| Feature | Description |
|---|---|
| Report Embedding | Display MultiQC HTML reports inline |
| Interactive Navigation | Navigate between MultiQC sections |
| Tool Integration | Support for various QC tools (FastQC, Samtools, STAR, etc.) |
Configuration¶
| Option | Description |
|---|---|
| Report Path | Path to MultiQC report HTML file |
| Display Mode | Full report or specific sections |
Data Requirements
MultiQC components require pre-generated MultiQC reports. The report HTML files should be accessible via the configured data source. See Managing Data Collections from the viewer for the create / append / replace / clear lifecycle.
Image Components (v0.7.0+)¶
Image components display image galleries from S3/MinIO storage, with metadata filtering and thumbnail previews.
Features¶
| Feature | Description |
|---|---|
| Grid Layout | Configurable column layout for image thumbnails |
| Thumbnail Previews | Auto-generated thumbnails with configurable size |
| Metadata Filtering | Filter images using interactive components |
| S3 Integration | Serve images directly from S3/MinIO buckets |
| Click to Expand | View full-size images in a modal |
Configuration¶
| Option | Description | Default |
|---|---|---|
| Data Collection | Source table with image metadata | Required |
| Image Column | Column containing image paths | Required |
| S3 Base Folder | S3/MinIO bucket path prefix | Required |
| Thumbnail Size | Thumbnail height in pixels | 150 |
| Columns | Number of columns in grid | 4 |
| Max Images | Maximum images to display | 20 |
Data Collection Setup¶
Image components require a Table-type Data Collection with:
- A column containing relative image paths (e.g.,
image_path) - Optional metadata columns for filtering (e.g.,
category,quality_score)
Example CSV structure:
sample_id,image_path,category,quality_score
sample_001,images/sample_001.png,A,0.95
sample_002,images/sample_002.png,B,0.87
sample_003,images/sample_003.jpg,A,0.92
Project YAML Configuration¶
Configure the Image data collection in your project.yaml:
data_collections:
- id: "650a1b2c3d4e5f6a7b8c9d10"
data_collection_tag: "sample_images"
description: "Sample images with metadata"
config:
type: "Image"
metatype: "Images"
scan:
mode: single
scan_parameters:
filename: images_data.csv
dc_specific_properties:
# Table fields (required for delta table)
format: "csv"
columns_description:
sample_id: "Unique sample identifier"
image_path: "Relative path to image file"
category: "Sample category"
quality_score: "Quality score 0-1"
# Image-specific fields (mandatory)
image_column: "image_path"
# Image display properties (optional)
s3_base_folder: "s3://bucket-name/images/"
local_images_path: ./images
supported_formats: [".png", ".jpg", ".jpeg"]
thumbnail_size: 150
Dashboard YAML Configuration¶
Add an image component in your dashboard_lite.yaml:
components:
- tag: sample-gallery
component_type: image
workflow_tag: python/image_workflow
data_collection_tag: sample_images
image_column: image_path
s3_base_folder: "s3://bucket-name/images/"
thumbnail_size: 150
columns: 3
max_images: 9
Image Storage¶
Images can be stored and accessed in two ways:
Option 1: Images already on S3/MinIO
If your images are already uploaded to S3/MinIO, specify the location in your project configuration:
Option 2: Upload local images with depictio-cli (Recommended)
Use the depictio-cli run command to automatically upload images from a local directory to S3:
# In project.yaml - dc_specific_properties
local_images_path: ./images # Local path relative to project directory
# Run depictio-cli to sync project and upload images
depictio-cli run --project-dir /path/to/project
The CLI will:
- Read the
local_images_pathfrom your project configuration - Upload images to the configured S3 bucket
- Set the correct
s3_base_folderautomatically
Cross-DC Filtering
Image components support filtering via interactive components on the same Data Collection. Select samples using a MultiSelect filter, and the image gallery updates automatically.
Map Components (v0.8.0+)¶
Map components display geospatial data on interactive tile-based maps using Plotly Express (no API key required).
Declaring coordinates at the DC level
lat_column / lon_column can be set per-figure (below) or at the Data Collection level so every Map figure on that DC inherits them. Map is the first example of type-specific DC configuration; more visualization types will follow.
Coordinates Data Collection (v0.12.0+)¶
Map components require a Table DC that explicitly declares its latitude and longitude columns. The DCTableCoordinatesConfig variant — defined at depictio/models/models/data_collections_types/table_coordinates.py:6 — extends the standard Table DC with three fields:
| Field | Required | Type | Default | Description |
|---|---|---|---|---|
lat_column |
✓ | str | — | Column holding latitude values |
lon_column |
✓ | str | — | Column holding longitude values |
crs |
— | str | EPSG:4326 |
Coordinate reference system |
It inherits every standard Table DC capability (CSV / TSV / Parquet format, polars_kwargs, keep_columns, columns_description). The dc_type stays "table" — the variant is materialised at deserialisation when lat_column / lon_column are present. A model validator enforces that the two columns must differ.

Map Types¶
| Type | Function | Best For |
|---|---|---|
scatter_map |
Point markers at lat/lon coordinates | Sample locations, site maps |
density_map |
Heatmap overlay from point density | Concentration hotspots |
choropleth_map |
Colored polygon regions from GeoJSON | Per-country/region statistics |
Configuration Options¶
| Option | Applies To | Description |
|---|---|---|
lat_column / lon_column |
scatter, density | Columns with GPS coordinates (required) |
color_column |
scatter, choropleth | Column for color encoding |
size_column |
scatter | Column for marker size encoding |
hover_columns |
scatter, choropleth | Columns shown on hover tooltip |
map_style |
all | Tile style: open-street-map, carto-positron, carto-darkmatter |
opacity |
all | Marker/region opacity (0.0–1.0) |
default_zoom / default_center |
all | Override auto-computed viewport |
Choropleth GeoJSON Sources¶
Choropleth maps require a GeoJSON FeatureCollection to define region boundaries. Three sources are supported:
| Source | Field | Description |
|---|---|---|
| URL | geojson_url |
Public URL to a GeoJSON file |
| Data Collection | geojson_dc_tag |
Tag of a geojson-type Data Collection in the same project |
| Inline | geojson_data |
Embedded GeoJSON dict (large — prefer URL or DC) |
Selection Filtering¶
Scatter maps support the same lasso/box/click selection as scatter plots. Enable with selection_enabled: true and selection_column. See Interactive Selection Filtering for details.
Choropleth Limitation
Choropleth maps do not support selection filtering (Plotly does not expose click/lasso on choropleth traces).
YAML Examples¶
Scatter map:
- tag: sampling-map
component_type: map
workflow_tag: python/my_workflow
data_collection_tag: sample_metadata
lat_column: latitude
lon_column: longitude
color_column: biome
size_column: read_count
hover_columns: [sample_id, site_name, country]
map_style: carto-positron
selection_enabled: true
selection_column: sample_id
Choropleth map:
- tag: country-choropleth
component_type: map
workflow_tag: python/my_workflow
data_collection_tag: sample_metadata
map_type: choropleth_map
locations_column: country_name
featureidkey: properties.NAME
color_column: sample_id
choropleth_aggregation: count
color_continuous_scale: Viridis
geojson_url: "https://raw.githubusercontent.com/nvkelso/natural-earth-vector/master/geojson/ne_110m_admin_0_countries.geojson"
Cross-DC Filtering¶
Interactive components can filter across linked Data Collections using the Links system. See Cross-DC Filtering for details.
┌─────────────────┐ ┌─────────────────┐
│ Filter: Status │ link │ Table: Samples │
│ [MultiSelect] │────────▶│ [auto-filters] │
└─────────────────┘ └─────────────────┘
Component Creation¶
Using the Stepper Wizard¶
The component builder guides you through creation:
- Select Data Collection - Choose your data source
- Choose Component Type - Figure, Table, Card, Interactive, Text, MultiQC, or Map
- Configure Settings - Type-specific options
- Preview - See the component before adding
- Add to Dashboard - Place on the canvas
Positioning Components¶
- Drag and drop components in Edit Mode
- Components snap to a grid layout
- Resize by dragging corner handles WATCHER-VERIFY-1778951276