nf-core/viralrecon¶
nf-core pipeline · nf-co.re/viralrecon
The viralrecon template covers the main outputs of a standard nf-core/viralrecon run:
- MultiQC quality control — FastQC, Cutadapt, samtools/picard alignment metrics
- Variant calling — iVar variants with gene, effect, and allele-frequency annotations (illumina only)
- Lineage assignment — Pangolin lineages with conflict and QC scores
- Clade assignment — Nextclade clades with substitution counts
- Coverage analysis — Mosdepth amplicon coverage, genome coverage, and amplicon heatmap
- Cross-sample landscape — variant landscape and lineage analysis dashboards
Works beyond SARS-CoV-2
The pipeline supports any viral genome in nf-core's reference-genomes config. This template was validated on SARS-CoV-2 / ARTIC amplicon data, but the recipe / dashboard structure carries over to other viruses with the same iVar variant-calling + Pangolin / Nextclade lineage layout.
Quick start¶
viralrecon needs no extra template variables — the same command works for
both sequencing platforms, which Depictio auto-detects from the run's
params.json:
Full dashboard: MultiQC, coverage & depth, lineage & clustering, variants, sample QC.
IS_NANOPORE is auto-detected: the coverage and lineage collections are
repointed at the artic_minion/ layout and the illumina-only variant
collections are dropped — see Conditional routes in the Reference.
--variant_caller ivar is required
The viralrecon template's recipes hardcode paths under variants/ivar/
(see variants_long.py, pangolin_lineages.py, nextclade_results.py).
Running nf-core/viralrecon with the alternative --variant_caller bcftools
produces a different output layout that the template won't match.
Aggregated data collections
The viralrecon DCs use metatype: "Aggregated". They are built
by recipes that fan multiple per-sample files into a single delta
table via glob_pattern. See Recipes
for the underlying mechanism.
Reference¶
Recipe DCs fan per-sample files into one delta table via glob_pattern; the
IS_NANOPORE route (auto-detected from params.json) repoints
coverage/lineage DCs at the artic_minion/ layout and drops the
illumina-only variant DCs.
Template variables¶
Variables you provide when running the template — DATA_ROOT via --data-root, the rest via --var NAME=value:
| Variable | Required | Description |
|---|---|---|
DATA_ROOT |
✓ | Root directory containing viralrecon pipeline output (multiqc/, variants/) |
Auto-detected (set from the run's metadata / params.json; the route flags drive Conditional routes below): IS_NANOPORE
Data collections¶
14 data collections — 2 required 12 optional.
| Tag | Type | Source | Recipe / scan target | Status |
|---|---|---|---|---|
multiqc_data |
MultiQC | scan | multiqc/multiqc_data/multiqc.parquet |
required |
summary_metrics |
Table | transformed | multiqc/summary_metrics.py |
required |
variants_long |
Table | transformed | ivar/variants_long.py |
optional |
pangolin_lineages |
Table | transformed | pangolin/pangolin_lineages.py |
optional |
nextclade_results |
Table | transformed | nextclade/nextclade_results.py |
optional |
mosdepth_amplicon_coverage |
Table | scan | variants/bowtie2/mosdepth/amplicon/all_samples.mosdepth.coverage.tsv |
optional |
mosdepth_genome_coverage |
Table | scan | variants/bowtie2/mosdepth/genome/all_samples.mosdepth.coverage.tsv |
optional |
mosdepth_amplicon_heatmap |
Table | scan | variants/bowtie2/mosdepth/amplicon/all_samples.mosdepth.heatmap.tsv |
optional |
oncoplot_canonical |
Table | transformed | nf-core/viralrecon/oncoplot_canonical.py |
optional |
complex_heatmap_canonical |
Table | transformed | mosdepth/complex_heatmap_canonical.py |
optional |
coverage_track_canonical |
Table | transformed | mosdepth/coverage_track_canonical.py |
optional |
sankey_canonical |
Table | transformed | nf-core/viralrecon/sankey_canonical.py |
optional |
upset_canonical |
Table | transformed | nf-core/viralrecon/upset_canonical.py |
optional |
variant_feature_matrix_canonical |
Table | transformed | nf-core/viralrecon/variant_feature_matrix_canonical.py |
optional |
Conditional routes¶
Rows are data collections; columns are the variables you set or params.json flags auto-detected from the run. Each filled cell is the effect of setting that variable; an empty cell means that variable leaves the collection unchanged. (4 collections are unaffected by any variable — present on every run.)
+ included− removed⇄ repointed
| Data collection | IS_NANOPORE |
|---|---|
summary_metrics | − |
variants_long | − |
pangolin_lineages | ⇄ |
nextclade_results | ⇄ |
mosdepth_amplicon_coverage | ⇄ |
mosdepth_genome_coverage | ⇄ |
mosdepth_amplicon_heatmap | ⇄ |
oncoplot_canonical | − |
upset_canonical | − |
variant_feature_matrix_canonical | − |
Cross-DC links¶
7 links — selecting a value in the source collection filters the target. The join column is shown after the source.
| Source · column | Target | Filters | |
|---|---|---|---|
summary_metrics · sample | → | multiqc_data | Filter MultiQC by sample selections from summary metrics |
summary_metrics · sample | → | variants_long | Filter variants table by selected samples |
summary_metrics · sample | → | pangolin_lineages | Filter Pangolin lineages by selected samples |
summary_metrics · sample | → | nextclade_results | Filter Nextclade results by selected samples |
summary_metrics · sample | → | mosdepth_amplicon_coverage | Filter amplicon coverage by selected samples |
summary_metrics · sample | → | mosdepth_genome_coverage | Filter genome coverage by selected samples |
summary_metrics · sample | → | mosdepth_amplicon_heatmap | Filter amplicon heatmap by selected samples |
Recipes¶
Each recipe reshapes raw pipeline output into a tidy table. The name links to its source; Output lists the validated EXPECTED_SCHEMA columns.
| Recipe | Transforms | Output |
|---|---|---|
ivar/variants_long.py |
Clean and normalize viralrecon variants_long_table.csv for dashboard consumption. | sample, CHROM, POS, REF, ALT, FILTER, DP, REF_DP, ALT_DP, AF, GENE, AA, EFFECT, FUNCLASS, mutation_label |
mosdepth/complex_heatmap_canonical.py |
Canonical-schema ComplexHeatmap DC for viralrecon amplicon coverage. | sample |
mosdepth/coverage_track_canonical.py |
Canonical-schema Coverage Track DC for viralrecon. | chromosome, position, value |
multiqc/summary_metrics.py |
Parse viralrecon summary_variants_metrics_mqc.csv into a clean per-sample metrics table. | sample, num_reads_mapped, pct_reads_mapped, coverage_median, pct_genome_covered_1x, pct_genome_covered_10x, num_variants_snp, num_variants_indel, num_variants_total, lineage |
nextclade/nextclade_results.py |
Extract and clean Nextclade clade assignment results from viralrecon output. | sample, clade, Nextclade_pango, totalSubstitutions, totalDeletions, totalInsertions, totalFrameShifts, totalMissing, totalNonACGTNs, alignmentScore, coverage, qc_overallScore, qc_overallStatus |
nf-core/viralrecon/oncoplot_canonical.py |
Canonical-schema Oncoplot DC for viralrecon variants. | sample_id, gene, mutation_type |
nf-core/viralrecon/sankey_canonical.py |
Canonical-schema Sankey DC for viralrecon lineage / clade typing. | sample, qc_status, lineage, clade |
nf-core/viralrecon/upset_canonical.py |
Canonical-schema UpSet DC for viralrecon variants. | mutation_label |
nf-core/viralrecon/variant_feature_matrix_canonical.py |
Canonical-schema sample × variant feature matrix for live PCA embedding. | sample_id |
pangolin/pangolin_lineages.py |
Extract and clean Pangolin lineage assignments from viralrecon output. | sample, lineage, conflict, ambiguity_score, scorpio_call, scorpio_support, pangolin_version, qc_status |
Dashboard tabs¶
The viralrecon template ships a five-tab dashboard (MultiQC parent +
four child tabs). Each tab targets a different analytical question;
filters propagate across tabs via cross-DC links on the
summary_metrics.sample column.
Pipeline-level quality control powered by MultiQC.
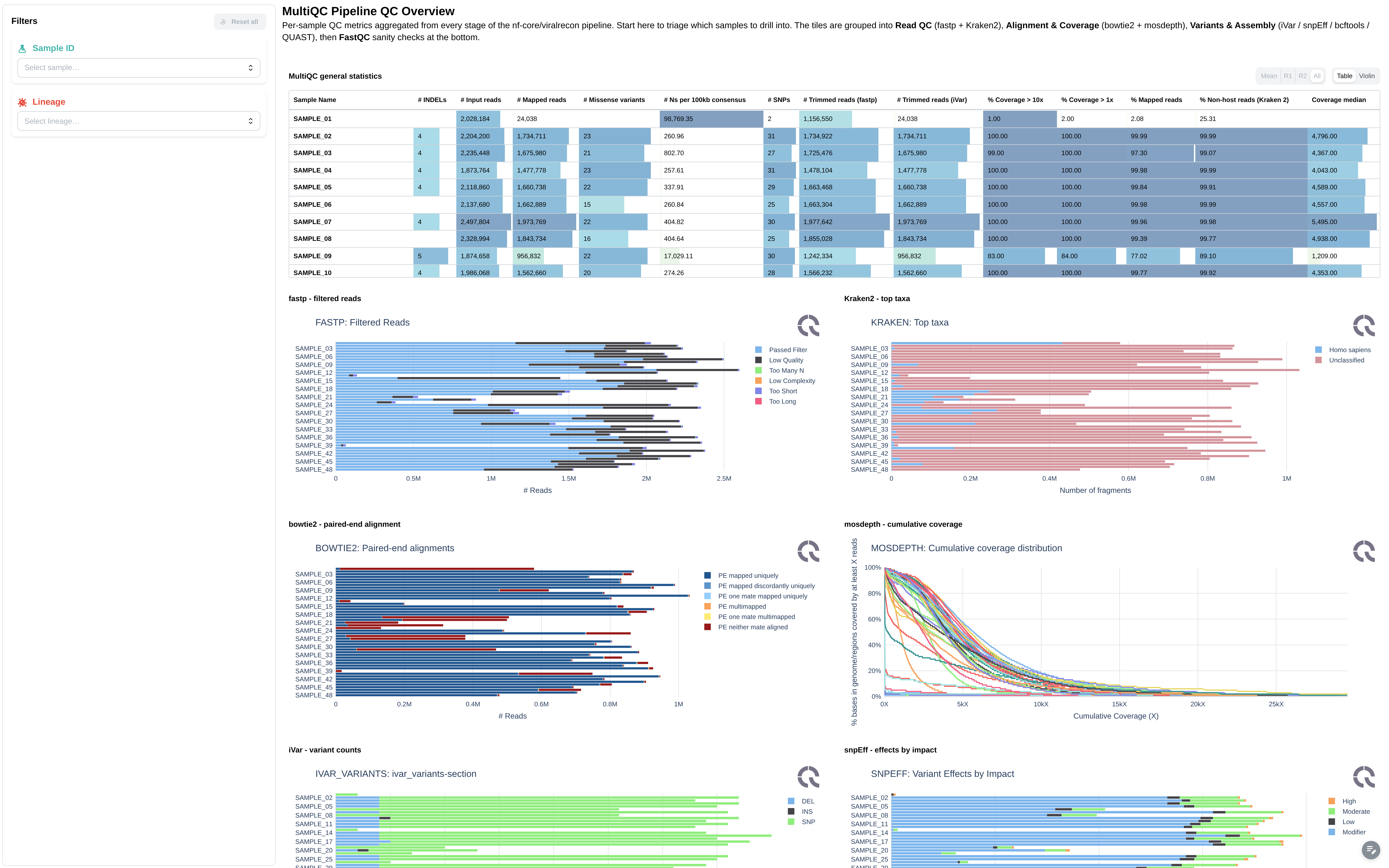
Filters: Sample ID, Lineage.
Components:
- General stats table
- Raw read counts and trimming statistics (FastQC, Cutadapt)
- Alignment rate and duplication rate
- samtools / picard alignment metrics
- Per-sample variant counts
Per-sample and per-amplicon coverage view.
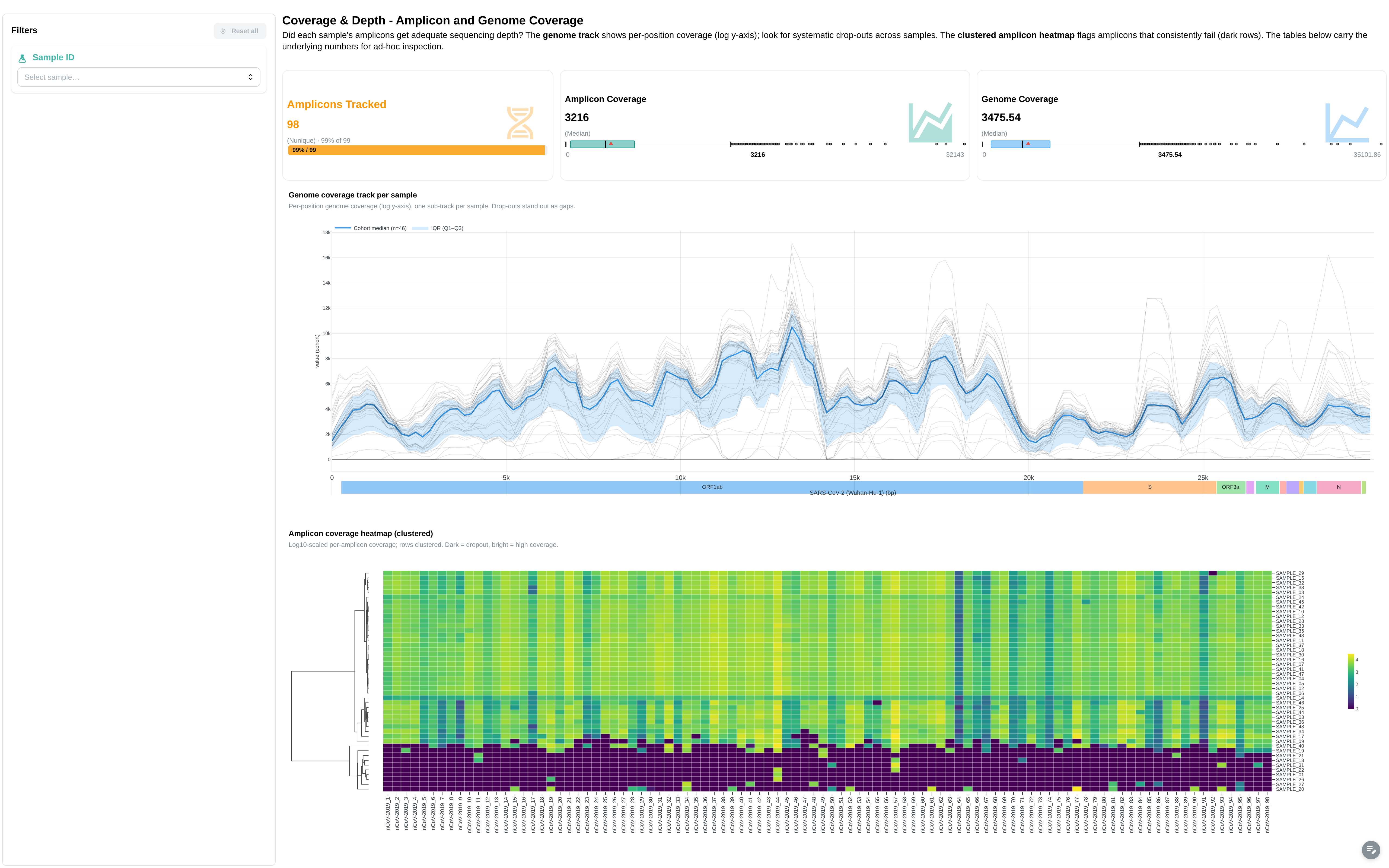
Filters: Sample ID.
Components:
- 4 summary cards: Total Samples, Amplicons Tracked, Amplicon Coverage, Genome Coverage
- Genome Coverage per Sample (line chart)
- Amplicon Coverage Heatmap
- Amplicon Coverage Data table
- Genome Coverage Data table
Pangolin lineage and Nextclade clade assignment, plus a Sankey funnel from QC status → lineage → clade.
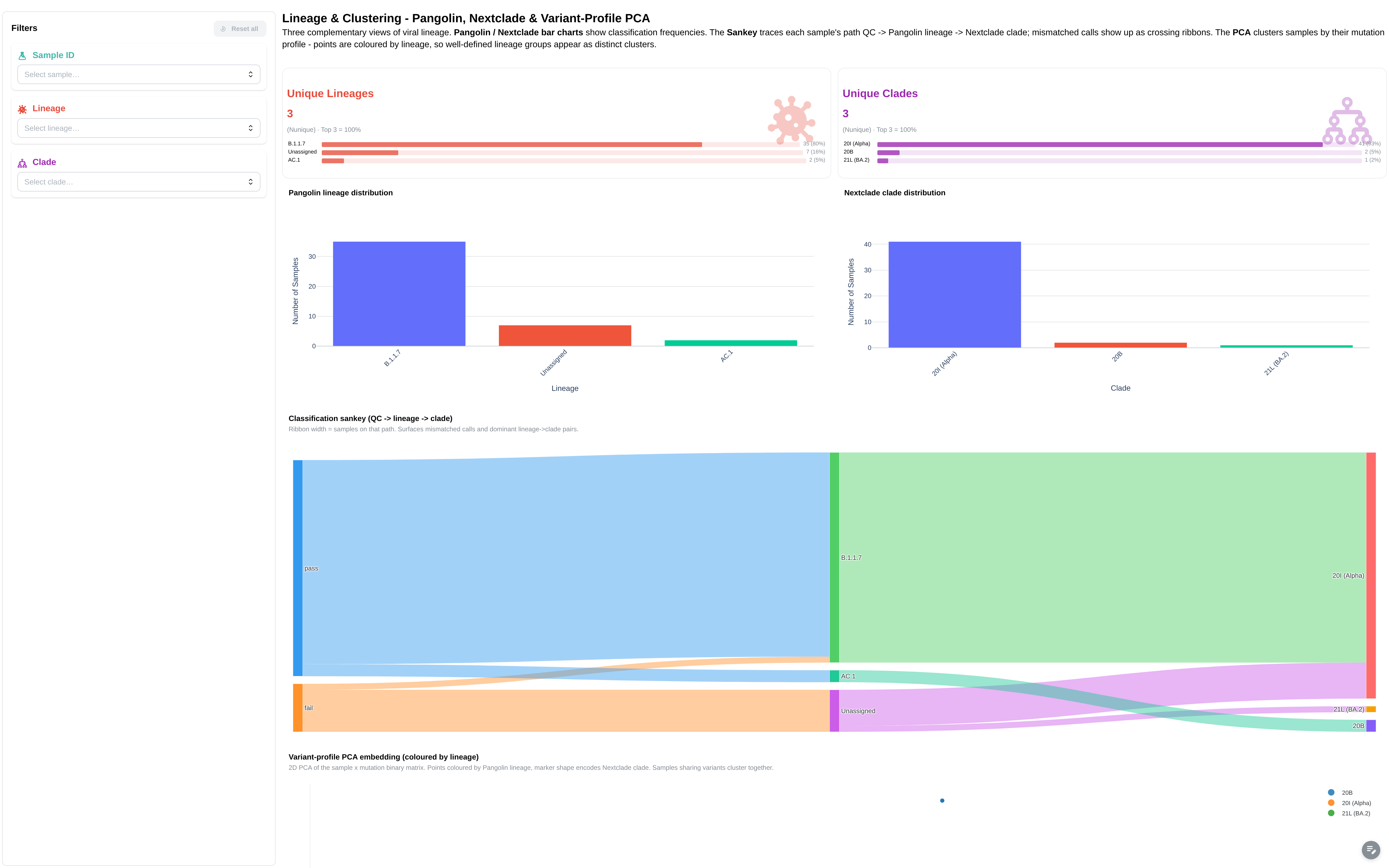
Filters: Sample ID, Lineage, Clade, QC Status.
Components:
- 4 summary cards: Total Samples, Unique Lineages, Unique Clades, Avg Genome Coverage (10x)
- 6 figures: Pangolin Lineage Distribution, Nextclade QC Status Overview, Nextclade Clade Distribution, Coverage vs Total Variants by Lineage, Genome Coverage per Sample (>= 10x Depth), Nextclade — Substitutions vs Deletions by Clade
- Sankey funnel: qc_status → lineage → clade (canonical sankey)
- 3 tables: Pangolin Lineage Assignments, Nextclade Clade Assignments, Summary Metrics
Variant calls and functional effects, with manhattan-style genome landscape and oncoplot of high-impact mutations.
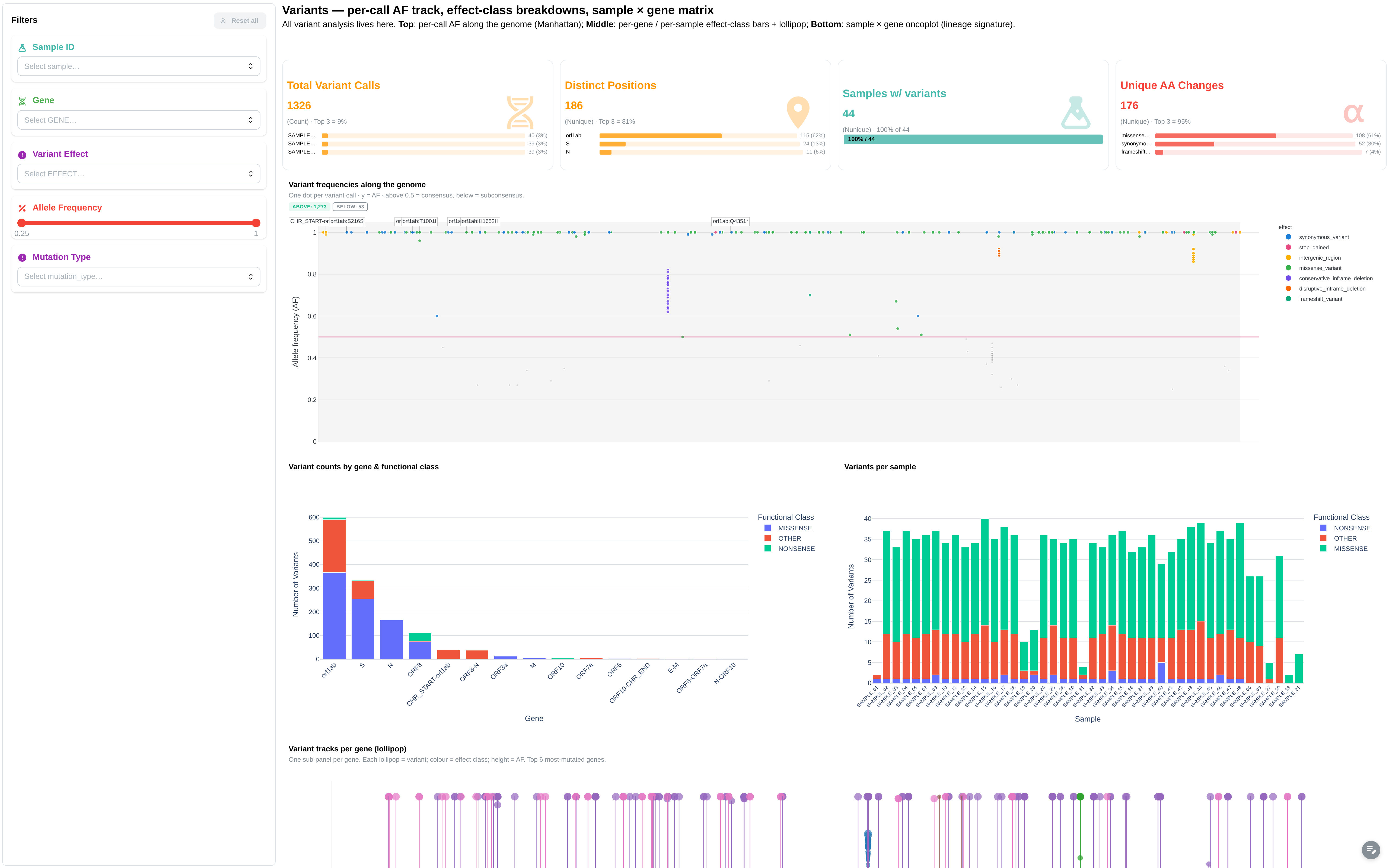
Filters: Sample ID, Gene, Variant Effect, Functional Class, Allele Frequency (range), Read Depth (range).
Components:
- 4 summary cards: Total Variants, Unique Genes, Mean Allele Freq, Unique AA Changes
- Manhattan plot: chr × pos × score (canonical manhattan)
- Lollipop: per-gene variants (canonical lollipop)
- Oncoplot: sample × gene × mutation_type (canonical oncoplot)
- 5 figures: Allele Frequency vs Genome Position, Variant Count by Gene and Functional Class, Variant Effect Distribution, Variant Functional Class Distribution, Variant Count per Sample
- 1 table: Variants Long Table
Per-sample QC scorecard combining alignment, coverage, variant counts and lineage / clade assignment in one place.
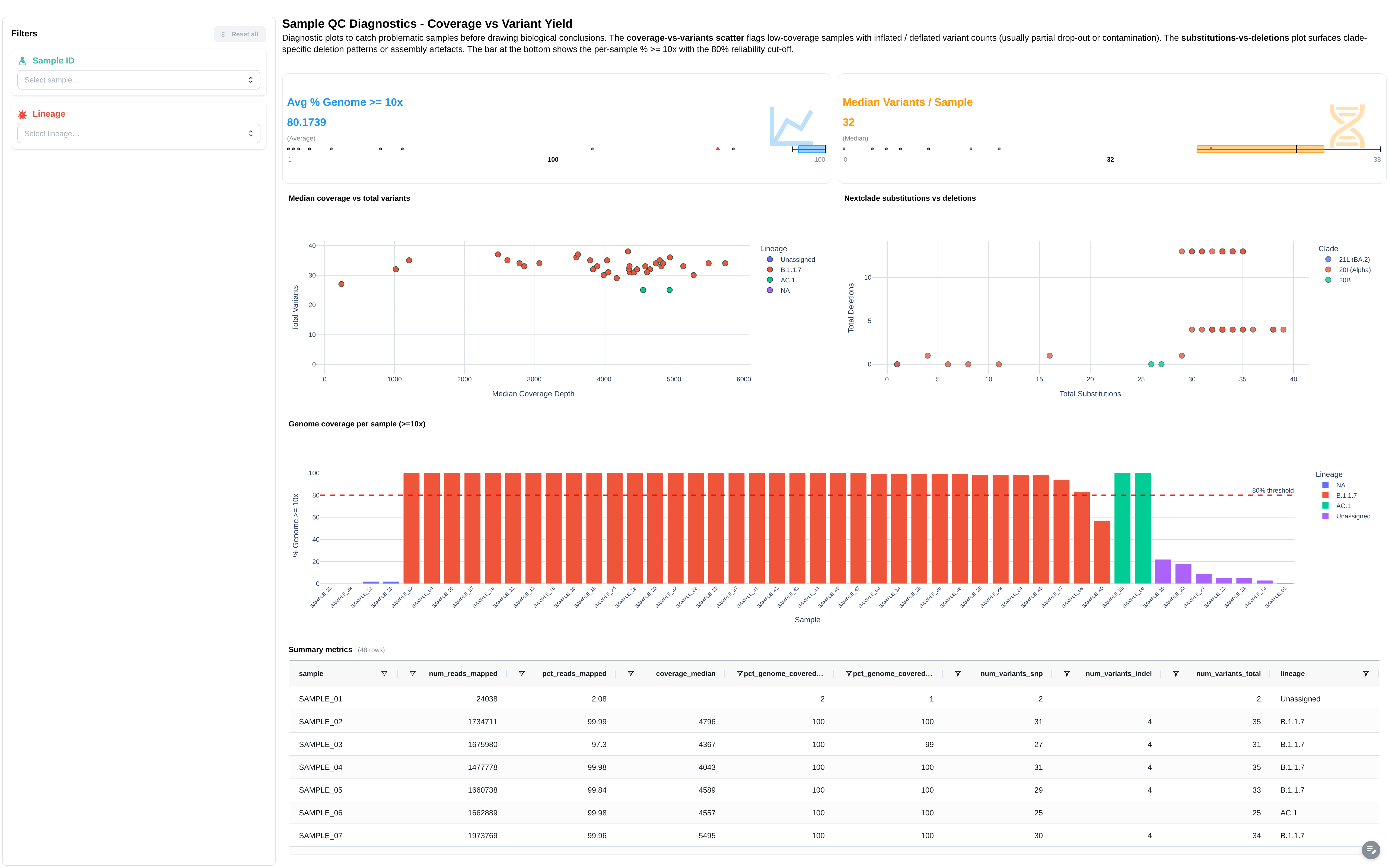
Filters: Sample ID, Lineage, QC Status.
Components:
- Summary cards: total samples, samples passing QC, mean coverage, mean variants per sample
- Sample × metric heatmap (canonical complex heatmap)
- Summary metrics table
Running the pipeline¶
Depictio reads the output of nf-core/viralrecon — it does not run the pipeline. Run the pipeline first, using the iVar variant caller the template targets:
nextflow run nf-core/viralrecon -r 3.0.0 \
--input samplesheet.csv \
--platform illumina \
--protocol amplicon \
--variant_caller ivar \
-profile docker
Then point Depictio at the results:
See nf-co.re/viralrecon/usage for full pipeline documentation.
Required data structure¶
Point --data-root to the directory containing your viralrecon outputs. This can be a single run's results/ folder or a parent directory containing multiple runs — Depictio scans recursively. Not all files are required; the template adapts to what's present and to the sequencing platform (IS_NANOPORE is auto-detected from the run's params.json).
<DATA_ROOT>/
├── multiqc/
│ ├── multiqc_data/
│ │ └── multiqc.parquet
│ └── summary_variants_metrics_mqc.csv
└── variants/
└── ivar/ # illumina layout (⚠ artic_minion/ on nanopore)
├── consensus/
│ └── bcftools/
│ ├── pangolin/*.pangolin.csv # Pangolin lineage, one file per sample
│ └── nextclade/*.csv # Nextclade clade, one file per sample
├── variants_long_table.csv # ⚠ illumina only (dropped on nanopore)
└── *.mosdepth.{coverage,heatmap}.tsv # amplicon / genome coverage
Test data¶
A small test fixture is available for local development without re-running
the full pipeline. The repository ships
download_test_data.sh
which fetches a real viralrecon run from nf-core's AWS megatest bucket:
bash depictio/projects/nf-core/viralrecon/3.0.0/download_test_data.sh \
--target /tmp/viralrecon_test
This pulls a published run from
s3://nf-core-awsmegatests/viralrecon/results-395079f1d24dce731ac22e03d7a5e71f110103fc/
and validates that all expected file patterns are present.
Once the download finishes, run depictio against it:
Alternative: run nf-core/viralrecon locally
The script can also re-run nf-core/viralrecon end-to-end if you'd rather regenerate the fixture from scratch:
Additional resources¶
- nf-co.re/viralrecon — official pipeline documentation
- nf-co.re/viralrecon/3.0.0/results — AWS test results
- Template System Reference — YAML format, variables, conditionals
- Recipes — how to read, test, and write recipes