nf-core/ampliseq¶
nf-core pipeline · nf-co.re/ampliseq
The ampliseq template covers the main outputs of a standard nf-core/ampliseq run:
- MultiQC quality control — FastQC read quality, Cutadapt trimming statistics
- Taxonomy composition — phylum-level barplots, sunburst, heatmap with annotations
- Alpha diversity — Faith's Phylogenetic Diversity, rarefaction curves (requires metadata)
- Differential abundance — ANCOM-BC volcano plots, log-fold change (requires metadata +
--ancombc) - Sampling locations — geographic scatter map from metadata coordinates (requires metadata)
Quick start¶
Reference¶
Running without METADATA_FILE prunes the metadata-dependent collections
(see the Conditional routes table); the --skip_qiime / --skip_taxonomy
/ multi-region routes are auto-detected from the run's params.json.
Template variables¶
Variables you provide when running the template — DATA_ROOT via --data-root, the rest via --var NAME=value:
| Variable | Required | Description |
|---|---|---|
DATA_ROOT |
✓ | Root directory containing ampliseq pipeline output (multiqc/, qiime2/) |
SAMPLESHEET_FILE |
— | Path to the ampliseq samplesheet. Auto-detected from {DATA_ROOT}/input/ (CSV or TSV) when omitted. |
METADATA_FILE |
— | Path to sample metadata TSV (--metadata in ampliseq run). Optional. All non-ID columns become annotations. |
METADATA_ID_COL |
— | Metadata sample-ID column name (links join on it). Auto-detected as the first metadata column; defaults to 'sample'. |
GROUP_COL |
— | Metadata column for grouping/facetting in dashboards. Auto-detected as first annotation column if not provided. |
Auto-detected (set from the run's metadata / params.json; the route flags drive Conditional routes below): GROUP_COL_DISPLAY, ANNOTATION_COLS, IS_MULTIREGION, SKIP_TAXONOMY, SKIP_ALPHA_RAREFACTION, SKIP_ANCOM
Data collections¶
23 data collections — 11 required 12 optional.
| Tag | Type | Source | Recipe / scan target | Status |
|---|---|---|---|---|
multiqc_data |
MultiQC | scan | multiqc/multiqc_data/multiqc.parquet |
required |
samplesheet |
Table | scan | {SAMPLESHEET_FILE} |
required |
metadata |
Table | scan | {METADATA_FILE} |
optional |
alpha_rarefaction |
Table | transformed | qiime2/alpha_rarefaction.py |
required |
taxonomy_composition |
Table | transformed | qiime2/taxonomy_composition.py |
required |
taxonomy_rel_abundance |
Table | transformed | nf-core/ampliseq/taxonomy_rel_abundance.py |
required |
sintax_rel_abundance |
Table | transformed | nf-core/ampliseq/sintax_rel_abundance.py |
optional |
taxonomy_heatmap |
Table | transformed | qiime2/taxonomy_heatmap.py |
required |
ancombc_results |
Table | transformed | qiime2/ancombc.py |
optional |
stacked_taxonomy_canonical |
Table | transformed | qiime2/stacked_taxonomy_canonical.py |
optional |
embedding_pcoa |
Table | transformed | qiime2/embedding_pcoa.py |
required |
rarefaction_canonical |
Table | transformed | qiime2/rarefaction_canonical.py |
optional |
alpha_diversity_multi_canonical |
Table | transformed | qiime2/alpha_diversity_multi_canonical.py |
optional |
complex_heatmap_canonical |
Table | transformed | nf-core/ampliseq/complex_heatmap_canonical.py |
required |
sunburst_canonical |
Table | transformed | nf-core/ampliseq/sunburst_canonical.py |
optional |
sankey_canonical |
Table | transformed | nf-core/ampliseq/sankey_canonical.py |
optional |
upset_canonical |
Table | transformed | nf-core/ampliseq/upset_canonical.py |
required |
ma_canonical |
Table | transformed | nf-core/ampliseq/ma_canonical.py |
required |
bray_curtis_canonical |
Table | transformed | nf-core/ampliseq/bray_curtis_canonical.py |
required |
phylogenetic_tree_canonical |
phylogeny | scan | {DATA_ROOT}/qiime2/phylogenetic_tree/tree.nwk |
optional |
phylogenetic_tree_metadata_canonical |
Table | transformed | nf-core/ampliseq/tree_metadata_canonical.py |
optional |
sidle_reconstructed |
Table | transformed | nf-core/ampliseq/sidle_reconstructed.py |
optional |
sidle_reconstruction_qc |
Table | transformed | nf-core/ampliseq/sidle_reconstruction_qc.py |
optional |
Conditional routes¶
Rows are data collections; columns are the variables you set or params.json flags auto-detected from the run. Each filled cell is the effect of setting that variable; an empty cell means that variable leaves the collection unchanged. (4 collections are unaffected by any variable — present on every run.)
+ included− removed⇄ repointed
| Data collection | METADATA_FILE | SKIP_QIIME | IS_MULTIREGION | SKIP_TAXONOMY | SKIP_ALPHA_RAREFACTION | SKIP_ANCOM |
|---|---|---|---|---|---|---|
metadata | + | |||||
alpha_rarefaction | + | − | − | − | ||
taxonomy_composition | − | − | ||||
taxonomy_rel_abundance | − | − | − | |||
sintax_rel_abundance | + | |||||
taxonomy_heatmap | − | − | − | |||
ancombc_results | + | − | − | − | − | |
stacked_taxonomy_canonical | − | − | − | |||
embedding_pcoa | − | − | − | |||
rarefaction_canonical | − | − | − | |||
alpha_diversity_multi_canonical | − | − | − | |||
complex_heatmap_canonical | − | − | − | |||
sunburst_canonical | − | − | − | |||
sankey_canonical | − | − | − | |||
upset_canonical | + | − | − | − | ||
ma_canonical | + | − | − | − | − | |
bray_curtis_canonical | − | − | − | |||
phylogenetic_tree_canonical | − | − | ||||
phylogenetic_tree_metadata_canonical | − | − |
Cross-DC links¶
18 links — selecting a value in the source collection filters the target. The join column is shown after the source.
| Source · column | Target | Filters | |
|---|---|---|---|
samplesheet · sampleID | → | multiqc_data | Filter MultiQC by samplesheet samples |
metadata · {METADATA_ID_COL} | → | multiqc_data | Filter MultiQC by metadata samples |
metadata · {METADATA_ID_COL} | → | alpha_rarefaction | Filter alpha rarefaction by metadata samples |
metadata · {METADATA_ID_COL} | → | taxonomy_composition | Filter taxonomy composition by metadata samples |
metadata · {METADATA_ID_COL} | → | taxonomy_rel_abundance | Filter taxonomy rel abundance by metadata samples |
samplesheet · sampleID | → | sintax_rel_abundance | Filter sintax rel abundance by samplesheet samples |
metadata · {METADATA_ID_COL} | → | sintax_rel_abundance | Filter sintax rel abundance by metadata samples |
samplesheet · sampleID | → | taxonomy_heatmap | Filter taxonomy heatmap by samplesheet samples |
metadata · {METADATA_ID_COL} | → | taxonomy_heatmap | Filter taxonomy heatmap by metadata samples |
metadata · {METADATA_ID_COL} | → | alpha_diversity_multi_canonical | Filter per-sample multi-metric alpha-diversity by metadata samples |
metadata · {METADATA_ID_COL} | → | rarefaction_canonical | Filter multi-metric rarefaction curves by metadata samples |
alpha_diversity_multi_canonical · sample_id | → | rarefaction_canonical | Filter rarefaction by Alpha Diversity tab sample selection |
metadata · {METADATA_ID_COL} | → | stacked_taxonomy_canonical | Filter stacked-taxonomy bars by metadata samples |
metadata · {METADATA_ID_COL} | → | embedding_pcoa | Filter PCoA embedding by metadata samples |
metadata · {METADATA_ID_COL} | → | sunburst_canonical | Filter sunburst by metadata samples |
metadata · {METADATA_ID_COL} | → | sankey_canonical | Filter sankey flows by metadata samples |
metadata · {METADATA_ID_COL} | → | complex_heatmap_canonical | Filter complex heatmap by metadata samples (best-effort on wide matrix) |
metadata · {METADATA_ID_COL} | → | bray_curtis_canonical | Filter Bray-Curtis distances by metadata samples (best-effort on wide matrix) |
Recipes¶
Each recipe reshapes raw pipeline output into a tidy table. The name links to its source; Output lists the validated EXPECTED_SCHEMA columns.
| Recipe | Transforms | Output |
|---|---|---|
nf-core/ampliseq/bray_curtis_canonical.py |
Canonical-schema Bray-Curtis distance DC for ampliseq. | sample |
nf-core/ampliseq/complex_heatmap_canonical.py |
Canonical-schema ComplexHeatmap DC for ampliseq. | Phylum, Kingdom |
nf-core/ampliseq/ma_canonical.py |
Canonical-schema MA-plot DC for ampliseq. | feature_id, avg_log_intensity, log2_fold_change |
nf-core/ampliseq/sankey_canonical.py |
Canonical-schema Sankey DC for ampliseq. | sample_id, Kingdom, Phylum, abundance |
nf-core/ampliseq/sidle_reconstructed.py |
Melt the SIDLE cross-region reconstructed feature table to long per-sample taxonomy. | feature_id, sample, count, taxonomy, Kingdom, Phylum, Class, Genus |
nf-core/ampliseq/sidle_reconstruction_qc.py |
Per-reconstructed-feature SIDLE reconstruction confidence (route-specific QC). | feature_id, num_regions, total_kmers_mapped, mean_kmer_per_region, stdv_kmer_per_region, mapped_asvs |
nf-core/ampliseq/sintax_rel_abundance.py |
Per-sample Phylum-level relative abundance from sintax (--skip_qiime) outputs. | sample, taxonomy, rel_abundance, Kingdom, Phylum |
nf-core/ampliseq/sunburst_canonical.py |
Canonical-schema sunburst DC for ampliseq. | Kingdom, Phylum, abundance |
nf-core/ampliseq/taxonomy_rel_abundance.py |
Transform QIIME2 relative abundance table to long-format per-sample taxonomy table. | sample, taxonomy, rel_abundance, Kingdom, Phylum |
nf-core/ampliseq/tree_metadata_canonical.py |
Tip metadata for the ampliseq phylogenetic tree. | taxon |
nf-core/ampliseq/upset_canonical.py |
Canonical-schema UpSet DC for ampliseq. | taxon |
qiime2/alpha_diversity_multi_canonical.py |
Per-sample multi-metric alpha-diversity DC for ampliseq. | sample_id, habitat |
qiime2/alpha_rarefaction.py |
Transform QIIME2 alpha rarefaction wide CSV to long-format rarefaction curves. | sample, depth, iter, faith_pd |
qiime2/ancombc.py |
Merge ANCOM-BC differential abundance results (5 files) into one long-format table. | id, contrast, lfc, p_val, q_val, w, se, Kingdom, Phylum, neg_log10_qval, significant |
qiime2/embedding_pcoa.py |
Canonical-schema embedding DC for ampliseq (PCoA on Bray-Curtis). | sample_id, dim_1, dim_2 |
qiime2/rarefaction_canonical.py |
Canonical-schema rarefaction DC for ampliseq (multi-metric). | sample_id, depth, iter, shannon, observed_features, faith_pd |
qiime2/stacked_taxonomy_canonical.py |
Canonical-schema stacked-taxonomy DC for ampliseq. | sample_id, taxon, rank, abundance |
qiime2/taxonomy_composition.py |
Transform QIIME2 barplot CSV (wide) to long-format taxonomy composition table. | sample, taxonomy, count |
qiime2/taxonomy_heatmap.py |
Pivot relative abundance table to wide-format heatmap matrix. | Phylum, Kingdom |
Dashboard tabs¶
The ampliseq dashboard ships as a six-tab funnel (MultiQC parent + five
child tabs). Filters propagate across tabs via cross-DC links on the
metadata sample column — see Cross-DC links in the
Reference section.
Quality control overview powered by MultiQC.

Filters: Sample ID, Habitat Type, Sampling Period (DatePicker).
Components:
- General stats table
- Cutadapt: filtered reads, trimmed sequence lengths
- FastQC: sequence counts, quality histograms, GC content, adapter content, status checks, Per-sequence quality / GC / N content, sequence duplication levels, length distribution
Within-sample diversity metrics, rarefaction, and per-habitat comparisons. Extended mode only.
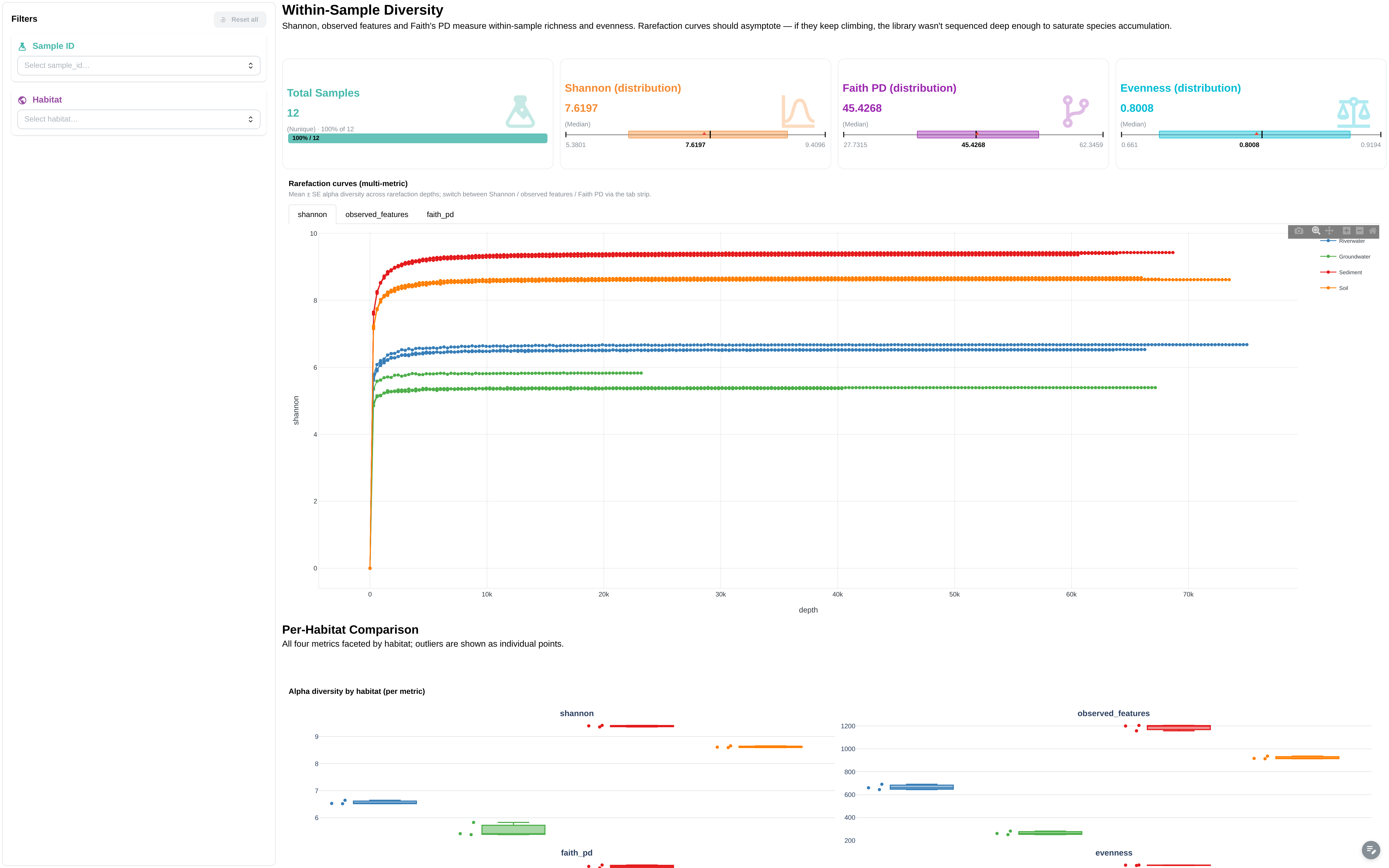
Filters: Sample ID, Habitat.
Components:
- 4 metric cards: Total Samples, Shannon (distribution), Faith PD (distribution), Evenness (distribution)
- Rarefaction curves (multi-metric) — advanced viz, filterable by habitat / sample via the in-tab DCLink
- Alpha diversity by habitat (per metric) — facetted boxplot
- Per-sample alpha diversity data table
Taxonomy composition + sampling-location map (extended mode).
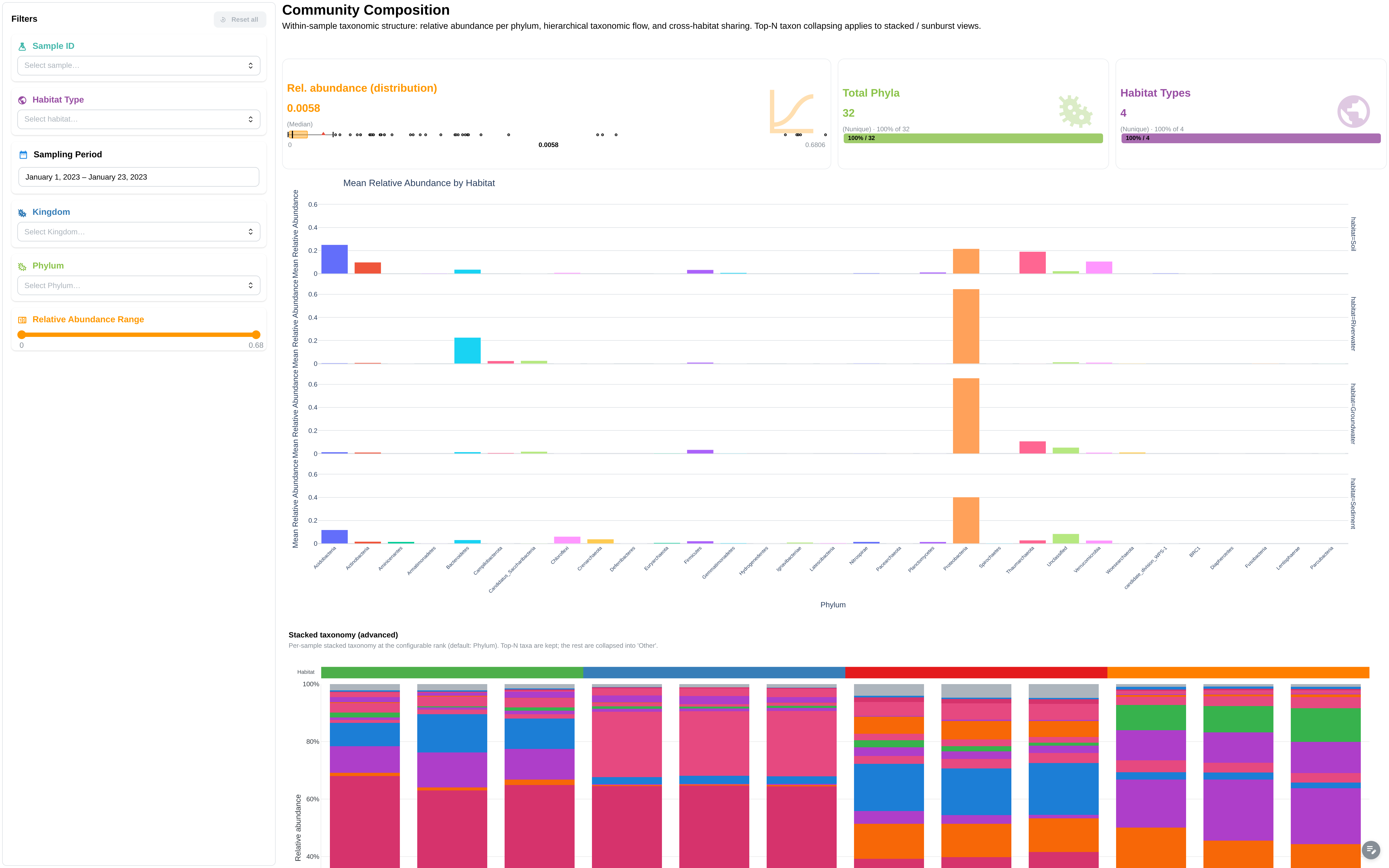
Components (base):
- Metric cards: total samples, total taxa, kingdoms, unique phyla
- Sunburst: Kingdom → Phylum hierarchy
- Mean relative abundance by Phylum (± std)
- Stacked bar: taxonomic composition per sample
- ComplexHeatmap: z-score normalized, clustered, with Kingdom row annotations
- Data table: taxonomy relative abundance
- Filters: Kingdom, Phylum, relative abundance range
Additional components (extended):
- Facetted bar charts by GROUP_COL
- Sampling locations scatter map
- Heatmap with habitat + city column annotations
- Filters: sampling period (DatePicker), GROUP_COL, sample ID
ANCOM-BC differential abundance results. Extended mode only.
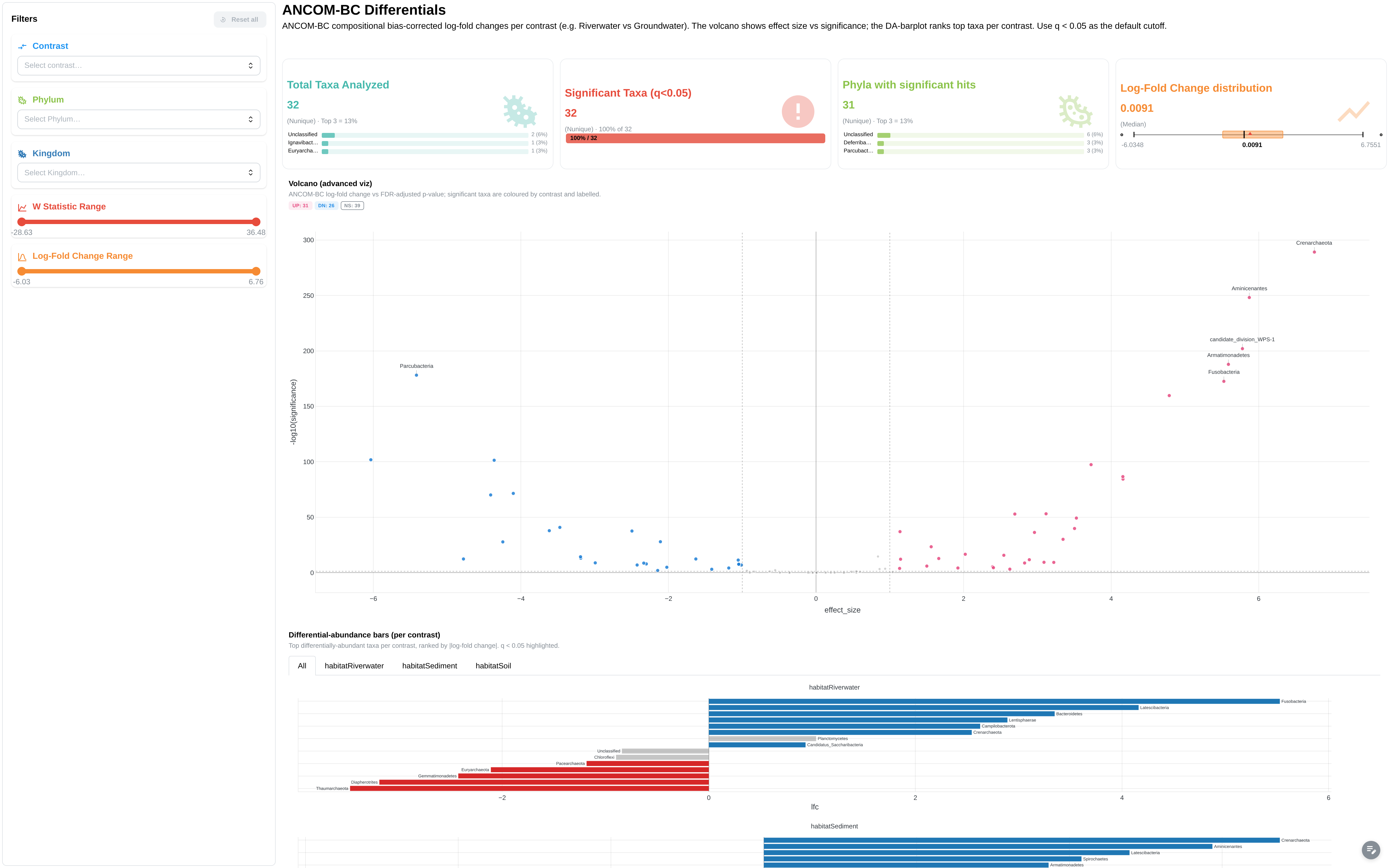
Components:
- Metric cards: total taxa, significant taxa (q<0.05), unique phyla, max log-fold change
- Volcano plot: LFC vs -log10(q-value), facetted by contrast
- DA barplot: per-contrast log-fold change
- Top differential taxa bar chart
- Results data table
- Filters: contrast, Phylum, Kingdom, W statistic range, LFC range
Beta-diversity / PCoA embedding + ComplexHeatmap on the canonical feature matrix. Surfaces clusters and outliers across samples.
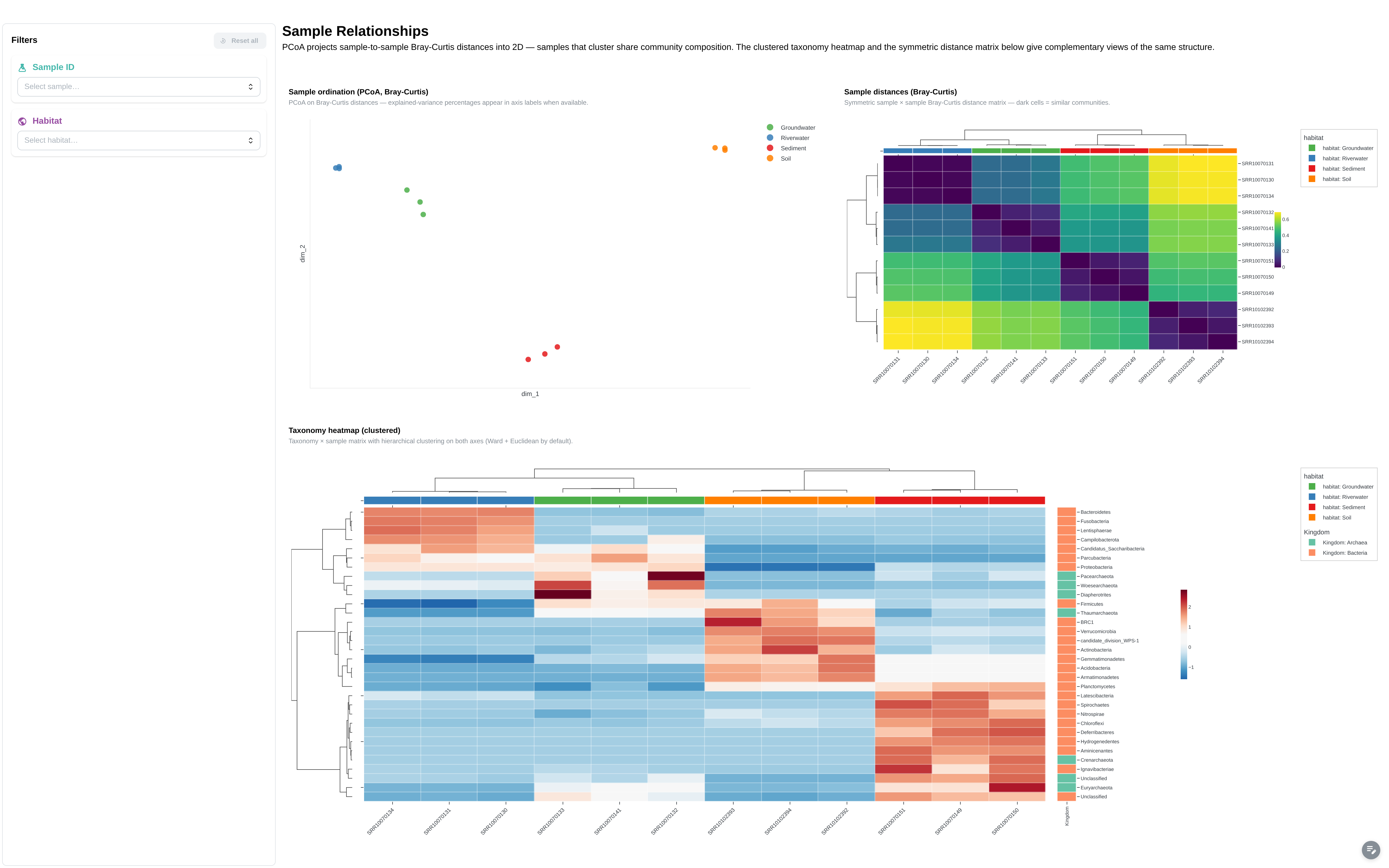
Components:
- Embedding (PCoA): 2D sample projection, colour-coded by habitat
- ComplexHeatmap: clustered z-score heatmap on the canonical feature matrix
- Bray-Curtis sample-distance heatmap
Rooted phylogenetic tree of ASVs with tip metadata overlay.
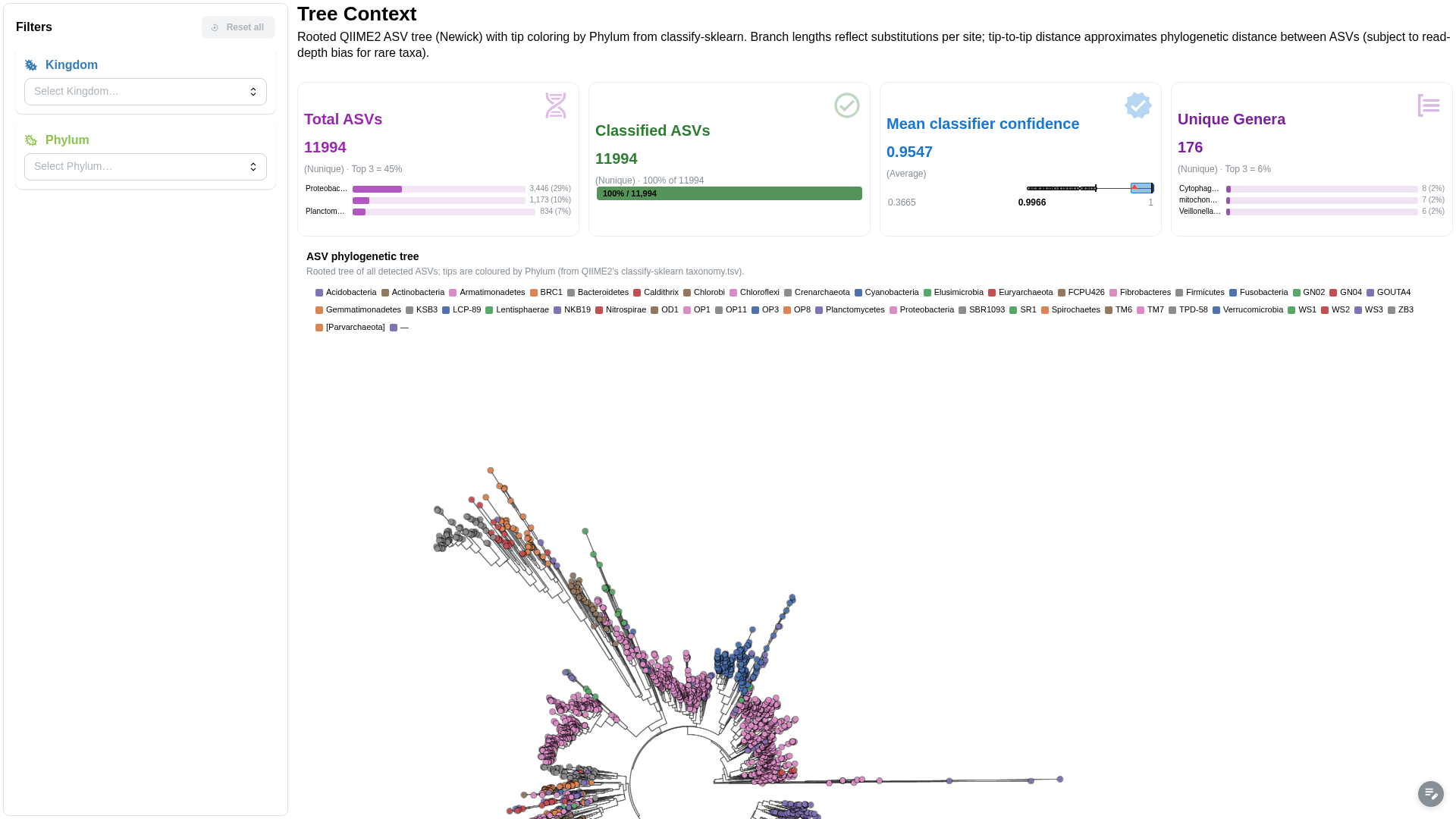
Components:
- Phylogenetic tree viewer (Newick) with metadata-annotated tips
Running the pipeline¶
Depictio reads the output of nf-core/ampliseq — it does not run the pipeline. Run the pipeline first:
nextflow run nf-core/ampliseq \
--input samplesheet.csv \
--FW_primer GTGYCAGCMGCCGCGGTAA \
--RV_primer GGACTACNVGGGTWTCTAAT \
--metadata Metadata.tsv \
-profile docker
Then point Depictio at the results:
depictio run --template nf-core/ampliseq/2.16.0 \
--data-root results/ \
--var SAMPLESHEET_FILE=samplesheet.csv \
--var METADATA_FILE=Metadata.tsv
See nf-co.re/ampliseq/usage for full pipeline documentation.
Required data structure¶
Point --data-root to the directory containing your ampliseq outputs. This can be a single run's results/ folder or a parent directory containing multiple runs — Depictio scans recursively. Not all files are required; the template adapts based on what's present and which --var flags you provide.
<DATA_ROOT>/
├── samplesheet.csv # --var SAMPLESHEET_FILE
├── Metadata.tsv # --var METADATA_FILE (optional)
└── <run_id>/ # One or more pipeline run output folders
├── multiqc/
│ └── multiqc_data/
│ └── multiqc.parquet
└── qiime2/
├── alpha-rarefaction/ # ⚠ Requires --metadata
│ └── faith_pd.csv
├── ancombc/differentials/ # ⚠ Requires --metadata + --ancombc
│ └── Category-<GROUP_COL>-level-2/
│ ├── lfc_slice.csv
│ ├── p_val_slice.csv
│ ├── q_val_slice.csv
│ ├── se_slice.csv
│ └── w_slice.csv
├── barplot/
│ └── level-2.csv
├── diversity/alpha_diversity/ # ⚠ Requires --metadata
│ └── faith_pd_vector/
│ └── metadata.tsv
└── rel_abundance_tables/
└── rel-table-2.tsv
Additional resources¶
- nf-co.re/ampliseq — official pipeline documentation
- nf-co.re/ampliseq/2.16.0/results — AWS test results
- Template System Reference — YAML format, variables, conditionals
- Recipes — how to read, test, and write recipes